
Editor's Introduction
Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model
Military veterans who have been exposed to blasts and athletes who have had multiple concussions often have neurological and cognitive problems. Medical researchers have found that these problems are linked to traumatic brain injury (TBI), which can occur when there is a significant impact to the head. Researchers have also found that both of these groups show evidence of chronic traumatic encephalopathy (CTE), a type of neurodegenerative disease that is hard to identify because it cannot be diagnosed until after a patient has died. To better understand what causes CTE, the authors of this study looked for similarities in the postmortem brains of blast-exposed military veterans and young athletes who had suffered multiple concussions. They also developed a mouse model of blast neurotrauma that replicates the damage seen in postmortem brains of these two groups and links them to learning and memory deficits. This study sheds light on how TBI and CTE are linked, and opens the door for the development of new diagnostic methods, treatments, and preventative measures.
Paper Details
Abstract
Blast exposure is associated with traumatic brain injury (TBI), neuropsychiatric symptoms, and long-term cognitive disability. We examined a case series of postmortem brains from U.S. military veterans exposed to blast and/or concussive injury. We found evidence of chronic traumatic encephalopathy (CTE), a tau protein–linked neurodegenerative disease, that was similar to the CTE neuropathology observed in young amateur American football players and a professional wrestler with histories of concussive injuries. We developed a blast neurotrauma mouse model that recapitulated CTE-linked neuropathology in wild-type C57BL/6 mice 2 weeks after exposure to a single blast. Blast-exposed mice demonstrated phosphorylated tauopathy, myelinated axonopathy, microvasculopathy, chronic neuroinflammation, and neurodegeneration in the absence of macroscopic tissue damage or hemorrhage. Blast exposure induced persistent hippocampal-dependent learning and memory deficits that persisted for at least 1 month and correlated with impaired axonal conduction and defective activity-dependent long-term potentiation of synaptic transmission. Intracerebral pressure recordings demonstrated that shock waves traversed the mouse brain with minimal change and without thoracic contributions. Kinematic analysis revealed blast-induced head oscillation at accelerations sufficient to cause brain injury. Head immobilization during blast exposure prevented blast-induced learning and memory deficits. The contribution of blast wind to injurious head acceleration may be a primary injury mechanism leading to blast-related TBI and CTE. These results identify common pathogenic determinants leading to CTE in blast-exposed military veterans and head-injured athletes and additionally provide mechanistic evidence linking blast exposure to persistent impairments in neurophysiological function, learning, and memory.
Report
INTRODUCTION
Blast exposure from conventional and improvised explosive devices (IEDs) affects combatants and civilians in conflict regions around the world (1–4). Individuals exposed to explosive blast are at increased risk for traumatic brain injury (TBI) (2, 5–15) that is often reported as mild (16cf., 17). Blast-related TBI represents a neuropsychiatric spectrum disorder that clinically overlaps with chronic traumatic encephalopathy (CTE), a progressive tau protein–linked neurodegenerative disease associated with repetitive concussive injury in athletes (18–21). Neuropathological hallmarks of CTE include widespread cortical foci of perivascular tau pathology, disseminated microgliosis and astrocytosis, myelinated axonopathy, and progressive neurodegeneration. Clinical symptoms of CTE include progressive affective lability, irritability, distractability, executive dysfunction, memory disturbances, suicidal ideation, and in advanced cases, cognitive deficits and dementia.
Blast exposure is a known precipitant of brain injury in animals (22–37) and humans (38–42) and has been linked to CTE neuropathology in a single case report by Omalu et al. (43). Despite growing awareness of blast-related TBI, the mechanisms of injury and biological basis underpinning blast neurotrauma and sequelae remain largely unknown and a matter of significant controversy. Given the overlap of clinical signs and symptoms in military personnel with blast-related TBI and athletes with concussion-related CTE, we hypothesized that common biomechanical and pathophysiological determinants may trigger development of CTE neuropathology and sequelae in both trauma settings. Here, we combine clinicopathological correlation analysis and controlled animal modeling studies to test this hypothesis.
Video. Dr. Lee Goldstein explains the research presented in this paper.
Source: brainlinemilitary.org
RESULTS
CTE neuropathology in blast-exposed military veterans and athletes with repetitive concussive injury
We performed comprehensive neuropathological analyses (table S1) of postmortem brains obtained from a case series of military veterans with known blast exposure and/or concussive injury (n = 4 males; ages 22 to 45 years; mean, 32.3 years). We compared these neuropathological analyses to those of brains from young amateur American football players and a professional wrestler with histories of repetitive concussive injury (n = 4 males; ages 17 to 27 years; mean, 20.8 years) and brains from normal controls of comparable ages without a history of blast exposure, concussive injury, or neurological disease (n = 4 males; ages 18 to 24 years; mean, 20.5 years). Case 1, a 45-year-old male U.S. military veteran with a single close-range IED blast exposure, experienced a state of disorientation without loss of consciousness that persisted for ~30 min after blast exposure. He subsequently developed headaches, irritability, difficulty sleeping and concentrating, and depression that continued until his death 2 years later from a ruptured basilar aneurysm. His medical history is notable for a remote history of concussion associated with a motor vehicle accident at age 8 years. Case 2, a 34-year-old male U.S. military veteran without a history of previous concussive injury, sustained two separate IED blast exposures 1 and 6 years before death. Both episodes resulted in loss of consciousness of indeterminate duration. He subsequently developed depression, short-term memory loss, word-finding difficulties, decreased concentration and attention, sleep disturbances, and executive function impairments. His neuropsychiatric symptoms persisted until death from aspiration pneumonia after ingestion of prescription analgesics. Case 3, a 22-year-old male U.S. military veteran with a single close-range IED blast exposure 2 years before death. He did not lose consciousness, but reported headache, dizziness, and fatigue that persisted for 24 hours after the blast. He subsequently developed daily headaches, memory loss, depression, and decreased attention and concentration. In the year before his death, he became increasingly violent and verbally abusive with frequent outbursts of anger and aggression. He was diagnosed with posttraumatic stress disorder (PTSD) 3 months before death from an intracerebral hemorrhage. His past history included 2 years of high school football and multiple concussions from fist fights. Case 4, a 28-year-old male U.S. military veteran with two combat deployments, was diagnosed with PTSD after his first deployment 3 years before death. His history was notable for multiple concussions as a civilian and in combat, but he was never exposed to blast. His first concussion occurred at age 12 after a bicycle accident with temporary loss of consciousness and pre/posttraumatic amnesia. At age 17, he experienced a concussion without loss of consciousness from helmet-to-helmet impact injury during football practice. At age 25, he sustained a third concussion during military deployment with temporary alteration in mental status without loss of consciousness. Four months later at age 26, he sustained a fourth concussion with temporary loss of consciousness and posttraumatic amnesia resulting from a motor vehicle–bicycle collision. Afterward, he experienced persistent anxiety, difficulty concentrating, word-finding difficulties, learning and memory impairment, reduced psychomotor speed, and exacerbation of PTSD symptoms. He died from a self-inflicted gunshot wound 2 years after his last concussion. The athlete group included Case 5, a 17-year-old male high school American football player who died from second impact syndrome 2 weeks after sustaining a concussion; Case 6, an 18-year-old high school American football and rugby player with a history of three to four previous concussions, one requiring hospitalization, who died 10 days after his last concussion; Case 7, a 21-year-old male college American football player, who played as a lineman and linebacker but had never been diagnosed with a concussion during his 13 seasons of play beginning at age 9, and who died from suicide; and Case 8, a 27-year-old male professional wrestler who experienced more than 9 concussions during his 10-year professional wrestling career who died from an overdose of OxyContin. The normal control group included Case 9, an 18-year-old male who died suddenly from a ruptured basilar aneurysm; Case 10, a 19-year-old male who died from a cardiac arrhythmia; Case 11, a 21-year-old male who died from suicide; and Case 12, a 24-year-old male who died from suicide.
Neuropathological analysis of postmortem brains from military veterans with blast exposure and/or concussive injury revealed CTE-linked neuropathology characterized by perivascular foci of tau-immunoreactive neurofibrillary tangles (NFTs) and glial tangles in the inferior frontal, dorsolateral frontal, parietal, and temporal cortices with predilection for sulcal depths (Fig. 1, A, B, E, F, and I to X). NFTs and dystrophic axons immunoreactive for monoclonal antibody CP-13 (Fig. 1, A to I, L, Q, R, and U, and fig. S1) directed against phosphorylated tau protein at Ser202 (pS202) and Thr205 (pT205), monoclonal antibody AT8 (Fig. 1S) directed against phosphorylated tau protein at Ser202 (pS202) and Thr205 (pT205), and monoclonal antibody Tau-46 (Fig. 1T) directed against phosphorylation-independent tau protein were detected in superficial layers of frontal and parietal cortex and anterior hippocampus. Evidence of axon degeneration, axon retraction bulbs, and axonal dystrophy were observed in the subcortical white matter subjacent to cortical tau pathology (Fig. 1, M and U to X, and fig. S1). Distorted axons and axon retraction bulbs were prominent in perivascular areas. Large clusters of LN3-immunoreactive activated microglia clusters (Fig. 1, K and P) were observed in subcortical white matter underlying focal tau pathology, but not in unaffected brain regions distant from tau lesions. Neuropathological comparison to brains from young-adult amateur American football players (Fig. 1, C, D, G, and H) with histories of repetitive concussive and subconcussive injury exhibited similar CTE neuropathology marked by perivascular NFTs and glial tangles with sulcal depth prominence in the dorsolateral and inferior frontal cortices. The young-adult athlete brains also revealed evidence of robust astrocytosis and multifocal axonopathy in subcortical white matter. Clusters of activated perivascular microglia were noted in the subcortical U-fibers. Neuropathological findings in the military veterans with blast exposure and/or concussive injury and young-adult athletes with repetitive concussive injury were consistent with our previous CTE case studies (20, 21) and could be readily differentiated from neuropathology associated with Alzheimer’s disease, frontotemporal dementia, and other age-related neurodegenerative disorders. Control sections omitting primary antibody demonstrated no immunoreactivity. By contrast, none of the brains from the four young-adult normal control subjects demonstrated phosphorylated tau pathology, axonal injury, subcortical astrocytosis, or microglial nodules indicative of CTE or other neurodegenerative disease (fig. S2).

Fig. 1. CTE neuropathology in postmortem brains from military veterans with blast exposure and/or concussive injury and young athletes with repetitive concussive injury. (A and E) Case 1, phosphorylated tau (CP-13) neuropathology with perivascular neurofibrillary degeneration in the frontal cortex of a 45-year-old male military veteran with a history of single close-range blast exposure 2 years before death and a remote history of concussion. Whole-mount section. Scale bar (E), 100 μm. (B and F) Case 2, phosphorylated tau (CP-13) neuropathology with perivascular neurofibrillary degeneration in the frontal cortex of a 34-year-old male military veteran with history of two blast exposures 1 and 6 years before death and without a history of concussion. Whole-mount section. Scale bar (F), 100 μm. (C and G) Case 6, phosphorylated tau (CP-13) neuropathology with perivascular neurofibrillary degeneration in the frontal cortex of an 18-year-old male amateur American football player with a history of repetitive concussive injury. Whole-mount section. Scale bar (G), 100 μm. (D and H) Case 7, phosphorylated tau (CP-13) neuropathology with perivascular neurofibrillary degeneration in the frontal cortex of a 21-year-old male amateur American football player with a history of repetitive subconcussive injury. Whole-mount section. Scale bar (H), 100 μm. (I) Case 1, phosphorylated tau (CP-13) immunostaining in the parietal cortex revealed a string of perivascular foci demonstrating intense immunoreactivity (areas enclosed by hash lines). Whole-mount section. (J) Case 1, phosphorylated neurofilament (SMI-34) immunostaining in adjacent parietal cortex section demonstrating colocalization of multifocal axonal swellings and axonal retraction bulbs surrounding small blood vessels (black circles) relative to perivascular tau foci (areas enclosed by hash lines). Whole-mount section. (K) Case 1, human leukocyte antigen–DR (HLA-DR) (LN3) immunostaining in adjacent parietal cortex section demonstrating colocalization of microglial clusters (black circles) relative to perivascular tau foci (areas enclosed by hash lines). Whole-mount section. (L) Case 1, high-magnification micrograph of phosphorylated tau (CP-13) immunostaining in the parietal cortex demonstrating string of perivascular phosphorylated tau foci. Whole-mount section. (M) Case 1, phosphorylated tau (PHF-1, brown) and phosphorylated neurofilament (SMI-34, red) double immunostaining in parietal cortex demonstrating axonal swellings and a retraction bulb (arrow) in continuity with phosphorylated tau neuritic abnormalities. Whole-mount section. Scale bar, 100 μm. (N) Case 1, phosphorylated neurofilament (SMI-34) immunostaining showing diffuse axonal degeneration and multifocal irregular axonal swellings in subcortical white matter subjacent to cortical tau pathology. Whole-mount section. (O) Case 1, phosphorylated neurofilament (SMI-34) immunostaining demonstrating perivascular axonal pathology and axonal retraction bulbs near a small cortical blood vessel. Whole-mount section. (P) Case 1, activated microglia (LN3) immunostaining showing a large microglial nodule in the subcortical white matter subjacent to cortical tau pathology. LN3 immunostaining was not observed in brain areas devoid of tau pathology. Whole-mount section. Scale bar, 100 μm. (Q) Case 2, phosphorylated tau (CP-13) immunostaining showing diffuse neuronal tau pathology (pre-tangles) in the hippocampal CA1 field. Whole-mount section. (R) Case 2, phosphorylated tau (CP-13) pathology in temporal cortex. Whole-mount section. (S) Case 1, phosphorylated tau (AT8) immunostaining showing diffuse neuronal tau pathology (pre-tangles) in the hippocampal CA1 field. Whole-mount section. (T) Case 1, phosphorylation-independent total tau (Tau-46) immunostaining in the frontal cortex. Whole-mount section. (U) Case 3, phosphorylated tau (CP-13) immunostained axonal varicosities in the external capsule of a 22-year-old male military veteran with a history of a single close-range IED blast exposure and remote history of concussions. Whole-mount section. (V to X) Case 3, SMI-34 immunostained axonal varicosities and retraction bulbs in the thalamic fasiculus and external capsule. Whole-mount sections
Major question
Do military veterans with a history of blast exposure develop the same CTE-linked brain abnormalities (pathologies) as athletes?
Overview
The panels in Figure 1 are sections of brain tissue that have been stained to detect various proteins. Panels A-I, L, and Q-S are stained for phosphorylated tau protein using one of two different antibodies. The antibodies used here are widely recognized as the standard for diagnosis of CTE.
Results
Using the same staining methods as in previous studies of CTE in athletes, the authors showed that the brains of blast-exposed military veterans have identical abnormalities.
Furthermore, the pathologies in both athletes and veterans are consistent with CTE. Both groups have abnormal accumulations of tau protein.
The patterns compared in this figure were chosen as the standard for diagnosis of CTE in the postmortem brain in the 2016 NIH consensus study.
Next questions
The authors found a correlation between CTE-linked brain abnormalities and previous military blast exposure or sports-related head injury. The next step was to determine whether that correlation was based on a causal linkage. They asked the following questions:
- Can blast exposure cause CTE (the case studies represent a limited population and can only suggest correlation)?
- Why is the CTE pathology conserved even though the injuries and backgrounds are different in each case?
Blast exposure induces traumatic head acceleration in a blast neurotrauma mouse model
We developed a murine blast neurotrauma model to investigate mechanistic linkage between blast exposure, CTE neuropathology, and neurobehavioral sequelae. Our compressed gas blast tube was designed to accommodate mice (fig. S3) and allowed free movement of the head and cervical spine to model typical conditions associated with military blast exposure (tables S2 and S3 and figs. S3 to S7). Wild-type C57BL/6 male mice (2.5 months) were anesthetized and exposed to a single blast with a static (incident) pressure profile comparable in amplitude, waveform shape, and impulse to detonation of 5.8 kg of trinitrotoluene (TNT) at a standoff distance of 5.5 m and in close agreement with ConWep (Conventional Weapons Effects Program) (Fig. 2A and table S3) (44). The model blast is comparable to a common IED fabricated from a 120-mm artillery round and is within the reported range of typical explosives, blast conditions, and standoff distances associated with military blast injury (45).

Fig. 2. Free-field pressure (FFP) and intracranial pressure (ICP) dynamics and head kinematics during single-blast exposure in a blast neurotrauma mouse model. (A) Measured incident static blast pressure (blue line) and blast impulse (red line) are compared to equivalent explosive blast waveform expected from 5.8 kg of TNT at a standoff distance of 5.5 m (black line) calculated according to software analysis using ConWep (44). The positive phase terminates at 4.8 ms (t+ = 4.8 ms; black hash line). Blast characteristics and waveform structure are comparable to a typical IED fabricated from a 120-mm artillery round (4.53 kg of TNT equivalent charge weight). The measured blast waveform and equivalent TNT blast waveform are in close agreement with a leading shock wavefront followed by a smooth decay. Note that ConWep presents an idealized blast resulting from an above-ground spherical charge and does not model negative-phase pressure transients or modulating factors commonly encountered in military blast scenarios. Reflecting surfaces, bounding structures (for example, crew compartments in armored vehicles, rooms within buildings, walled streets, and alleyways), local geometry, device and deployment characteristics (for example, encapsulation, internal reflectors, and open versus buried deployment), ambient environmental conditions, and other factors strongly influence blast pressure amplitude (positive and negative), phase duration, impulse history, waveform structure, and target interactions (30, 84–86). (B and C) ICP waveform and impulse profile in the brain of an intact living mouse (B) and isolated mouse head severed at the cervical spine (C) subjected to the same blast conditions as in (A). Blast waveforms recorded in the brains of living mice (B) and isolated heads (C) were similar in amplitude to each other and to the measured free-field static pressure. Small differences in the ICP signal waveforms were within the expected range given differences in frequency-dependent transducer response characteristics and experimental preparations. (D) Kinetographic representation of projected Cartesian motion of a representative mouse head during blast exposure as determined by high-speed videography acquired at 100,000 frames per second. Cartesian motion of the head was calculated by tracking a reflective paint mark on the snout. Labeled time points identify corresponding time points in (A) and (E) to (G). (E to G) Relative position (E), angular velocity (F), and angular acceleration (G) of the mouse head referenced to the horizontal (blue) and sagittal (red) planes of motion as determined by analysis of high-speed videographic records obtained during blast exposure. Head acceleration was most significant during the positive phase of the blast shock wave.
Overview
In Figure 1, the authors show that blast-exposed military veterans have CTE-linked brain abnormalities that are identical to the early-stage CTE pathology in young athletes with repetitive sports-related head injuries.
The authors' goal was to experimentally cause comparable brain abnormalities in mice using an accurate experimental blast exposure model. Such a model would open the door to investigate the biomechanics of blast-related injury and identify how blast exposure is linked to CTE neuropathology.
Major question (Panel A)
What experimental blast conditions cause brain abnormalities in mice similar to those seen in the brains of blast-exposed military veterans?
The authors took a different approach to this question than previous studies. They attempted to recreate in mice the brain abnormalities that are observed in the brains of blast-exposed veterans. Many studies prior to this one attempted to replicate blast exposure on a mouse scale.
The model used in this study causes neuropathology, neurological symptoms, physiological impairments, and cognitive deficits similar to those caused by blast exposure in humans.
Panels B-G
What happens to the head and the brain during blast exposure?
In panels B and C, the authors used a small needle hydrophone inserted into the brain to investigate the relationship between pressure changes inside and outside the head. They found that the pressure in the brain closely matched the outside pressure.
They also investigated a popular (but not experimentally validated) hypothesis known as the "water hammer effect." This hypothesis holds that sudden blast exposure causes an increase in pressure in the thorax (chest cavity), creating a pressure wave (or "water hammer") which exerts pressure on the brain and causes injury. The authors showed that decapitation--separating the head from the thorax and circulatory system--did not significantly decrease the pressure in the brain during blast exposure. This allowed them to rule out the "water hammer effect" as the primary cause of brain injury.
From this, they concluded that a different mechanism must be responsible for blast-related brain injury.
Panels D-G
Panels D through G show the results of tracking the motion of the head during blast exposure. The authors used high-speed video cameras to record head movement at 100,000 frames per second.
They found that blast wind caused the head to move violently from side to side (because of this, the authors named it the "bobblehead effect").
This finding implies that, even though the origin of the force may be different (i.e., a blast or sport-related impact), the cause of the observed brain pathologies is traumatic head acceleration, or the "bobblehead effect." The authors investigated this hypothesis further in Figure 7.
To investigate intracranial pressure (ICP) dynamics during blast exposure, we inserted a needle hydrophone into the hippocampus of living mice and monitored pressure dynamics during blast exposure. We detected blast wavefront arrival times in the brain that were indistinguishable from corresponding free-field pressure (FFP) measurements in air (Fig. 2B) and in close agreement with ConWep analysis of an equivalent TNT blast (Fig. 2A and table S3). To investigate possible thoracic contributions to blast-induced ICP transients, we evaluated pressure tracings in the hippocampus of intact living mice (Fig. 2B) and compared results to the same measurements obtained in isolated mouse heads severed at the cervical spine (Fig. 2C). Blast-induced pressure amplitudes in the two experimental preparations were comparable to each other and to the corresponding FFP measurements in air, after accounting for the addition of the dynamic pressure on the head. Small differences in the pressure waveforms were within the expected range given frequency-dependent response characteristics of the transducers and differences in the two experimental preparations. We did not detect delayed blast-induced ICP transients in either preparation over recording times up to 100 ms. These observations indicate that blast wavefront transmission in the mouse brain is mediated without significant contributions from thoracovascular or hydrodynamic mechanisms.
In our experimental system, the blast shock wave traveling at ~450 m/s encountered the left lateral surface of the mouse head first, then traversed the ~11-mm skull width (46) in ~24 μs. The pressure differential associated with this traversal has an insignificant effect on skull displacement due to the short time interval. For the remainder of the waveform duration, the static pressures at the lateral surfaces of the skull are virtually identical and the corresponding transient effects are negligible. The air-skull impedance mismatch creates a back-reflected air shock as well as a rapidly moving (≥1500 m/s) transmitted shock wave, the latter taking a maximum of ~7 μs to traverse the cranium and cranial contents. Although the reflected and transmitted shock waves are large (~2.5 times greater than the 77-kPa incident overpressure), the ~7-μs traversal time of the skull-brain transmitted wave is short enough to allow rapid equilibration across the skull. Thus, the head acts acoustically as a “lumped element” (47, 48). The only significant pressure term remaining is the ~19-kPa peak dynamic pressure generated by blast wind. We concluded that an ICP transducer in the brain parenchyma should measure pressure differentials that do not differ by more than 19 kPa from FFP values, at least beyond the initial 30 μs after blast arrival. This analysis was confirmed by experimental measurements (Fig. 2B). Only the initial rise of the blast wave has a short enough time scale to be affected by propagation effects in the head, a prediction confirmed by the longer rise time of the ICP compared to the static FFP waveforms (Fig. 2, B and C). The remaining waveform components evenly distribute through the brain with amplitude and shape that approximate the static FFP (Fig. 2A).
The blast wave had a measured Mach number of 1.26 ± 0.04 (fig. S6), from which the calculated blast wind velocity was 150 m/s (336 miles/hour). Kinematic analysis of high-speed videographic records of head movement during blast exposure confirmed rapid oscillating acceleration-deceleration of the head in the horizontal and sagittal planes of motion (Fig. 2, D to G, and video S1). We calculated peak average radial head acceleration of 954 ± 215 krad/s2(Fig. 2G), corresponding to 100.2 N exerted on the head during blast exposure. Peak angular and centripetal acceleration were most significant during the positive phase of the blast shock wave. No appreciable head acceleration was detected after ~8 ms.
Single-blast exposure induces CTE-linked neuropathology, ultrastructural pathology, and phosphorylated tau proteinopathy in a blast neurotrauma mouse model
We hypothesized that blast forces exerted on the skull would result in head acceleration-deceleration oscillation of sufficient intensity to induce persistent brain injury (“bobblehead effect”). To evaluate this hypothesis, we studied brains from mice euthanized 2 weeks after exposure to a single blast or sham blast. Gross examination of postmortem brains from both groups of mice was unremarkable and did not reveal macroscopic evidence of contusion, necrosis, hematoma, hemorrhage, or focal tissue damage (Fig. 3, A to F, and fig. S8). In contrast, brains from blast-exposed mice showed marked neuropathology by immunohistological analysis (Fig. 3, H, J, L, Q, N, S, and T). Blast-exposed brains exhibited robust reactive astrocytosis throughout the cerebral cortex, hippocampus, brainstem, internal capsule, cerebellum, and corticospinal tract (Fig. 3, H and T) that was not observed in brains from sham-blast control mice (Fig. 3, G and O). Brains from blast-exposed mice also exhibited enhanced somatodendritic phosphorylated tau CP-13 immunoreactivity in neurons in the superficial layers of the cerebral cortex (Fig. 3J) that was not observed in the brains of sham-blast control mice (Fig. 3I). The cerebral cortex and CA1 field of the hippocampus in the brains of blast-exposed mice were also notable for clusters of chromatolytic and pyknotic neurons with nuclear and cytoplasmic smudging and beaded, irregularly swollen dystrophic axons (Fig. 3, L and Q) that were not observed in the brains of sham-blast control mice (Fig. 3, K and P). Hippocampal CA1 neurons in blast-exposed mice were intensely Tau-46–immunoreactive (Fig. 3, N and S) compared to sham-blast controls (Fig. 3, M and R) and additionally showed evidence of frank neurodegeneration in the hippocampal CA1 and CA3 subfields and dentate gyrus (Fig. 4 and fig. S9). Activated perivascular microglia were observed throughout the brain in blast-exposed mice and were especially notable in the cerebellum (Fig. 3T; compared to control, Fig. 3O). Patchy loss of cerebellar Purkinje cells with empty baskets was also noted in blast-exposed mice (fig. S10H) but not in sham-blast control mice (fig. S10G). Examination of the cervical spinal cords of blast-exposed mice did not reveal evidence of motor neuron dropout or degeneration (fig. S10, A and B). However, blast-exposed mice did show decreased choline acetyltransferase immunoreactivity in motor neurons in the cervical cord (fig. S10D) and cranial nerve XII (fig. S10F) when compared to sham-blast controls (fig. S10, C and E), suggesting loss of central cholinergic inputs.

Fig. 3. Single-blast exposure induces CTE-like neuropathology in wild-type C57BL/6 mice. (A to F) Absence of macroscopic tissue damage (contusion, necrosis, hematoma, or hemorrhage) 1 day (A to C) or 2 weeks (D to F) after exposure to a single blast. Experimental blast conditions were compatible with 100% survival and full recovery of gross locomotor function. (G) Normal astrocytic glial fibrillary acidic protein (GFAP) immunoreactivity in a mouse brain 2 weeks after exposure to sham blast. Whole-mount sections. (H) Increased astrocytic GFAP immunoreactivity in the ipsilateral cortex (area enclosed by white hash line), bilateral thalamus (white asterisks), and bilateral hypothalamus (black asterisks) 2 weeks after single-blast exposure. Parenchymal atrophy with ventricular dilation was also observed (white arrowhead). Whole-mount sections. (I) Background phosphorylated tau (CP-13) immunostaining in superficial layers of the cerebral cortex 2 weeks after exposure to sham blast. (J) Phosphorylated tau (CP-13) immunostaining in superficial layers of the cerebral cortex 2 weeks after exposure to a single blast. Increased accumulation of phosphorylated tau in the brains of blast-exposed mice was confirmed by quantitative immunoblot analysis (Fig. 5). (K and P) Background phosphorylated neurofilament (SMI-31) immunostaining in the hippocampus 2 weeks after exposure to sham blast demonstrating normal-appearing CA1 pyramidal neurons with no detectable axonal pathology. (L and Q) Increased phosphorylated neurofilament (SMI-31) immunostaining in the hippocampus 2 weeks after exposure to single blast demonstrating pyknotic CA1 pyramidal neurons with nuclear smudging and injured axons with beaded, irregular swellings [arrowhead, (Q); enlargement shown in inset]. (M and R) Faint total tau (Tau-46) immunoreactivity in the soma and processes of pyramidal neurons in the hippocampal CA1 field 2 weeks after exposure to sham blast. (Nand S) Increased total tau (Tau-46) immunoreactivity in the soma and processes of pyramidal neurons [arrowheads, (S)] in the hippocampal CA1 field 2 weeks after exposure to single blast. Biochemical abnormalities in total tau expression in the brains of blast-exposed mice were confirmed by quantitative immunoblot analysis (Fig. 5). (O) Faint activated microglial [Ricinus communis agglutinin (RCA)] immunoreactivity in the cerebellum 2 weeks after exposure to sham blast. (T) Increased activated microglial RCA immunoreactivity in the cerebellum indicative of brisk microgliosis [arrowheads, (T)] 2 weeks after exposure to single blast.
Major question
Does the authors' mouse model of blast TBI accurately recreate CTE-linked brain pathologies like those seen in postmortem brains from blast-exposed military veterans?
This figure shows that blast exposure in our mouse model causes similar CTE-linked brain pathologies as those seen in postmortem brains from blast-exposed military veterans. This figure shows the results 2 weeks after exposure to a single blast or a sham blast (non-blast control). The authors used immunohistochemistry to look at abnormal accumulation of hyperphosphorylated tau and other relevant proteins that are linked to CTE (as observed in postmortem human brains from blast-exposed military veterans).
Data
The clinical diagnosis of CTE can only be made by examination of the brain postmortem. In this figure, the authors begin by determining whether or not their mouse model of blast neurotrauma recreates key features of CTE like those in human blast cases.
The antibodies used to stain the mouse brain are the same as those used in the human cases (i.e., CP-13, Tau-46, SMI-31, GFAP, RCA). Panels A-F show gross pathology (the brain as visible to the naked eye) in mouse brains 2 weeks after single blast exposure or sham (no injury control) exposure. The absence of gross pathology and presence of CTE-linked microscopic pathology after blast exposure is consistent with the idea of blast-related TBI as an "invisible injury." More accurately, the injury is "invisible" to the naked eye, but is certainly visible by microscopic examination.
Results
The authors compared the brain pathologies in the blast-exposed mice to those in the sham (non-blast-exposed) control mice.
Single blast exposure increased expression of abnormally hyperphosphorylated tau in the brains of blast-exposed mice two weeks post-exposure.
The authors also saw evidence of brain inflammation (neuroinflammation) and astrocytosis. The specific abnormalities and their anatomical distribution in the brains of blast-exposed mice strikingly resemble CTE-linked brain pathologies observed in postmortem human brains from blast-exposed military veterans (Fig 1).
Next questions
If a single blast exposure in mice triggers CTE brain pathology, what else can we learn about how blast exposure damages the brain? What else would be helpful to know about this apparent cause-effect relationship? What experiments might be helpful to confirm, extend, or most importantly, invalidate ("falsify") the conclusions drawn from these findings?
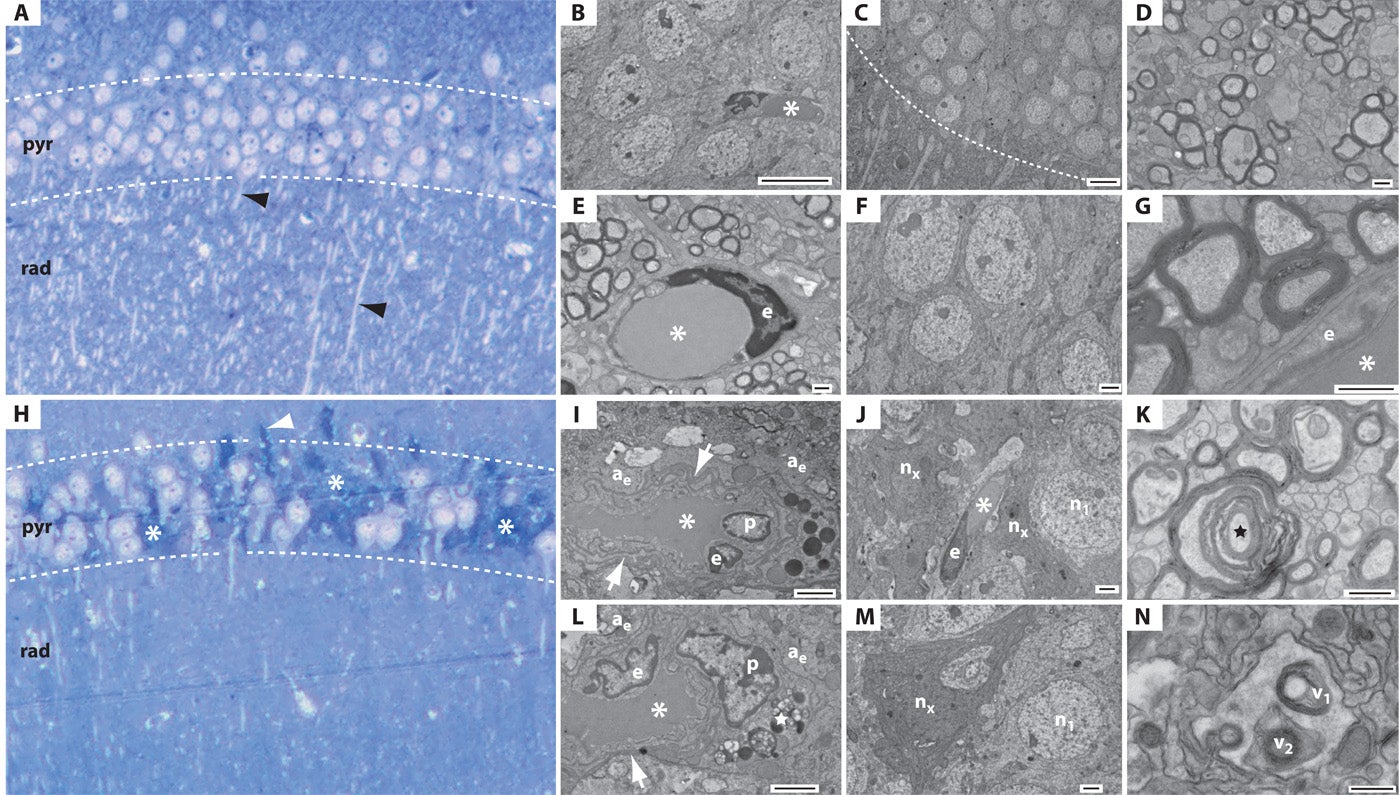
Fig. 4. Single-blast exposure induces hippocampal ultrastructural pathology in wild-type C57BL/6 mice. (A to G) Normal histology and ultrastructure in the hippocampal CA1 field 2 weeks after sham-blast exposure. (A) Toluidine blue–stained semithick section of the hippocampal CA1 field after sham blast. The CA1 field exhibits normal histological structure with a densely compacted layer of intact pyramidal neurons in the stratum pyramidale (pyr) and profuse dendritic profiles (black arrowheads) in the stratum radiatum (rad). (B to G) Electron micrographs of adjacent ultrathin sections demonstrating normal neuronal, axonal, and perivascular ultrastructure in the hippocampal CA1 field 2 weeks after sham-blast exposure. (B) CA1 pyramidal neurons in proximity to a capillary (asterisk) and endothelial cell. Scale bar, 10 μm. (C) Hippocampal CA1 field with normal stratum pyramidale (above white hash line) and stratum radiatum (below white hash line). Numerous dendrites are evident in the stratum radiatum. Scale bar, 10 μm. (D) Axon field in the stratum alveus demonstrating normal neuropil ultrastructure. Scale bar, 500 nm. (E) Capillary (asterisk) with endothelial cell nucleus (e) in a field of myelinated axons demonstrating normal ultrastructure in the stratum alveus. Scale bar, 500 nm. (F) Pyramidal neurons with normal ultrastructure in the hippocampal CA1 field. Scale bar, 2 μm. (G) Myelinated axons in transverse section in proximity to a capillary (asterisk) and endothelial cell (e). Scale bar, 500 nm. (H to N) Histological and ultrastructural pathology in the hippocampal CA1 field 2 weeks after single-blast exposure. (H) Toluidine blue–stained semithick section of hippocampus. Clusters of chromatolytic and pyknotic neurons (asterisks) are evident throughout the stratum pyramidale (pyr). Note the marked paucity of dendrites in the stratum radiatum (rad). A tortuous axon (white arrowhead) is present at the boundary between the stratum pyramidale and the stratum oriens. (I to N) Electron micrographs of adjacent ultrathin cryosections demonstrating widespread ultrastructural pathology in the hippocampal CA1 field 2 weeks after single-blast exposure. (I) Hydropic perivascular astrocytic end-feet (ae) surround an abnormal capillary (asterisk) and endothelial cell (e). The astrocytic end-feet are grossly distended and edematous. Numerous vacuoles are scattered throughout the pale cytoplasm. The capillary exhibits an abnormal shape and grossly thickened, tortuous basal lamina (white arrow). A pericyte (p) and numerous electron-dense inclusion bodies are also present. Scale bar, 2 μm. (J) Degenerating pyramidal neurons (nx) in proximity to a capillary (asterisk), endothelial cell (e), and swollen, hydropic processes of a perivascular astrocyte in the stratum pyramidale. A neighboring pyramidal neuron (n1) appears normal. Scale bar, 2 μm. An enlarged field of this same region is shown in fig. S20. (K) Degenerating myelinated nerve fiber (black star) in the stratum alveus. Scale bar, 500 nm. (L) Swollen, hydropic perivascular astrocyte end-feet (ae) surrounding a dysmorphic capillary (asterisk) in the hippocampal CA1 field. Note the abnormal endothelial cell (e) with irregularly shaped nucleus and nearby perivascular pericyte (p). The capillary basal lamina (white arrow) is grossly thickened. Lipofuscin granules (white star) are present in an adjacent process. Scale bar, 500 nm. A micrographic montage (fig. S11; corresponding high-magnification micrographs, fig. S12) of this same region reveals the soma and communicating processes of this perivascular astrocyte. (M) Degenerating CA1 pyramidal neuron (nx) in the stratum pyramidale of the hippocampal CA1 field. The electron-dense cytoplasm and condensed nucleus of this “dark neuron” correspond to the pyknotic neurons observed in toluidine blue–stained semithick sections (Fig. 4H). A neighboring neuron (n1) appears normal. Scale bar, 2 μm. (N) Presumptive autophagic vacuoles (v1, v2) in a perivascular astrocyte in the hippocampal CA1 field. Scale bar, 500 nm.
Major question
What happens to the fine structure of the brain two weeks after blast exposure?
The authors used an electron microscope to perform ultrastructural analysis of cellular and subcellular abnormalities. These abnormalities can't be seen with a regular light microscope.
Electron microscopy
Electorn microscopy uses beams of electrons for imaging, allowing a greater magnification than light microscopy. Electron microscopes can be used for imaging at atomic resolutions.
The microscope uses an electron beam, passing it through the sample. Materials that are more electron dense will interact more and appear darker.
Results
As with the light microscope pathology in Figure 3, electron microscopy revealed significant effects of blast exposure in blast-exposed mice.
The top collection of images, Panels A-G, are from a control mouse. The bottom collection, Panels H-N, are from a blast-exposed mouse.
Panels A and H show are semi-thick sections that provide a "bird's eye view" of the much thinner sections shown in Panels B-G and I-N. These sections are stained with Toluidine blue (to show neuronal cell bodies and axons) and were visualized with light microscopy. Panel A shows a sham control mouse and Panel H shows a mouse that was exposed to a blast two weeks earlier. These semi-thick sections are a reference to help provide context when viewing the much thinner sections in B-G and I-N.
Note the dramatic difference in appearance between the hippocampus of the control mouse (Panel A) and the blast-exposed mouse (Panel H).
In the control mouse, there is a layer of densely packed cells (Panel A, pyr), a healthy-appearing round capillary and capillary endothelial cells (Panel E), healthy neurons (Panels B, C, and F), and uniformly thick myelin sheaths wrapped around axons (Panels D and G).
In the blat-exposed mouse, there is a disrupted layer of pyrimidal cells (Panel H, pyr), an abnormally shaped capillary and endothelial cells (Panels I and L), abnormal neurons (Panels J and M), damaged myelin sheaths (Panel K), and evidence of autophagy (cellular "self-eating", Panel N).
The authors hypothesized that head acceleration during a blast causes forces that shear (tear) fine structures in the brain.
Next questions
Based on their observations of blast-induced damage to the structures and proteins in the brain at both the light microscope and electron microscope level, the authors decided to quantify these abnormalities in terms of CTE-related neurochemistry and neuronal function?
Ultrastructural pathology was observed in electron micrographs of neurons, axons, and capillaries in the hippocampi of blast-exposed mice but not in sham-blast control mice (Fig. 4and figs. S11 to S22). Examination of semithick sections of hippocampus CA1 and CA3 regions and dentate gyrus in brains from blast-exposed mice revealed clusters of chromatolytic and pyknotic neurons throughout the stratum pyramidale and a marked paucity of dendritic profiles in the stratum radiatum (Fig. 4H and fig. S9, B and C) that was not evident in the brains of sham-blast control mice (Fig. 4A and fig. S9A). Blast-related ultrastructural microvascular pathology was notable for the presence of hydropic perivascular astrocytic end-feet (Fig. 4, I and J, and figs. S11, S13 to S16, S19C, and S20). Pathologically swollen, edematous, and often highly vacuolated astrocytic end-feet were observed in association with dysmorphic capillaries marked by pathologically thickened, tortuous basal lamina and abnormal endothelial cells with irregularly shaped nuclei (Fig. 4L and figs. S11 to S16). Perivascular processes in the hippocampi of blast-exposed mice often contained inclusion bodies, lipofuscin granules, myelin figures, and autophagic vacuoles (Fig. 4, I, L, and N, and figs. S11, S12, S14, and S17 to S19). Pericytes (Fig. 4, I and L, and figs. S11, S12, S14, and S16), microglial cells (fig. S18), dystrophic myelinated nerve fibers (Fig. 4K and figs. S15 to S17 and S19A), and “dark neurons” (Fig. 4M and figs. S20 to S22) with electron-dense cytoplasm and irregularly shaped nuclei were frequently observed in proximity to these abnormal capillaries in blast-exposed mice. By contrast, the brains of sham-blast control mice exhibited normal hippocampal cytoarchitecture without evidence of ultrastructural neuropathology (Fig. 4, A to G).
To confirm the presence of phosphorylated tau proteinopathy in the brains of blast-exposed mice, we performed immunoblot analysis of tissue homogenates prepared from brains harvested from mice 2 weeks after single-blast or sham-blast exposure (Fig. 5). Immunoblot analysis revealed a significant blast-related elevation of phosphorylated tau protein epitopes pT181 and pS202 detected by monoclonal antibody CP-13 (Fig. 5, A, B, and G) and pT205detected by monoclonal antibody AT270 (Fig. 5, C, D, and I) that are associated with early neurodegenerative tau misprocessing. Blast-related tau phosphorylation was also detected when quantitated as a ratio of phosphorylated tau protein to total tau protein (Fig. 5, E, F, H, and J). In mice exposed to sham blast, all three of the major native murine tau isoforms (4R2N, 4R0N, and 4R1N) were evident (Fig. 5E). By contrast, immunoblots of brain homogenates prepared from mice exposed to a single blast revealed a tau protein isoform distribution pattern that was dominated by a single band corresponding to the intermediate-sized native tau isoform (4R1N; Fig. 5F). Phosphorylated tauopathy (Fig. 5, B and D) and tau isoform distribution abnormalities (Fig. 5F) were detected bilaterally, a finding consistent with blast-related CTE neuropathology and electrophysiological deficits. Blast-induced brain tau proteinopathy was confirmed by enzyme-linked immunosorbent assay (ELISA) analysis of tau protein phosphorylated at pSer199 (single blast, 40 ± 2 ng/ml; sham blast, 31 ± 2 ng/liter; P = 0.027, two-tailed Student’s t test).
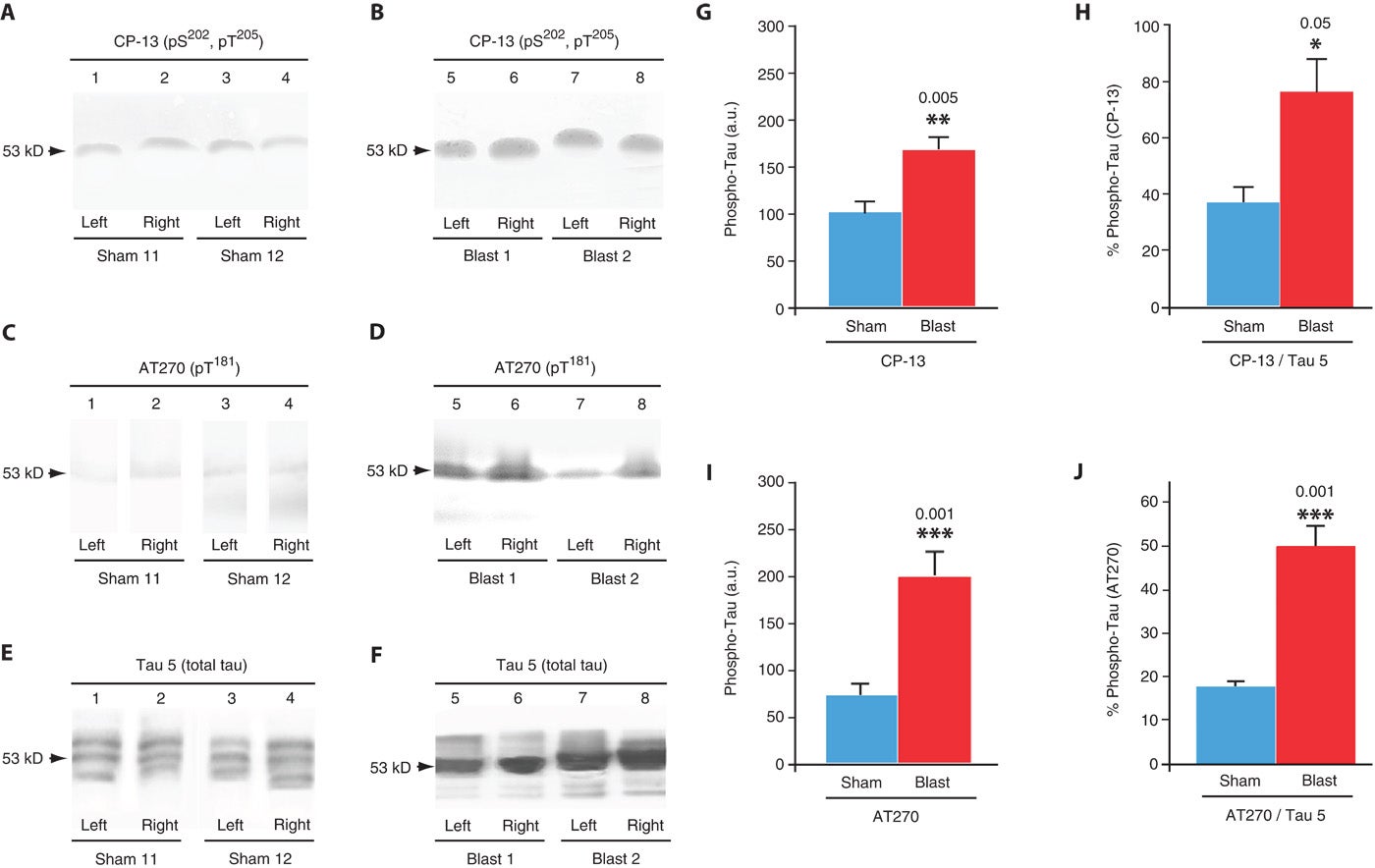
Fig. 5. Single-blast exposure induces increased brain tau protein phosphorylation in wild-type C57BL/6 mice. (A and B) Immunoblots of brain extracts from the left and right hemispheres of mice probed with monoclonal antibody CP-13 directed against phosphorylated tau protein (pS202/pT205) 2 weeks after exposure to sham blast (lanes 1 to 4) or single blast (lanes 5 to 8). Note the single broad band that migrated with an apparent molecular mass of 53 kD (arrows) in brains from mice in both groups. (C and D) Immunoblots of brain extracts from the left and right hemispheres of mice probed with monoclonal antibody AT270 directed against phosphorylated tau protein (pT181) using the same homogenates as in (A) and (B). (E and F) Immunoblots of brain extracts from the left and right hemispheres of mice probed with monoclonal antibody Tau 5 directed against total tau protein using the same homogenates as in (A) to (D). Unlike the results shown in the preceding panels, Tau 5 immunoblots revealed an apparent blast-related alteration in tau protein isoform distribution. (G) Densitometric quantitation of CP-13 phosphorylated tau protein (pS202/pT205) immunolabel in brain homogenates from mice exposed to single blast or sham blast 2 weeks before euthanizing. Mean values ± SEM in arbitrary densitometric units (a.u.). P < 0.005, two-tailed Student’s t test. (H) Densitometric quantitation of CP-13 phosphorylated tau protein (pS202/pT205) immunolabel in brain homogenates as a proportion of total tau protein (Tau 5) in brain homogenates from mice exposed to single blast or sham blast 2 weeks before euthanizing. Mean values ± SEM in arbitrary densitometric units. P < 0.05, two-tailed Student’s t test. (I) Densitometric quantitation of AT270 phosphorylated tau protein (pT181) immunolabel in brain homogenates from mice exposed to single blast or sham blast 2 weeks before euthanizing. Mean values ± SEM in arbitrary densitometric units. P < 0.001, two-tailed Student’s t test. (J) Densitometric quantitation of AT270 phosphorylated tau protein (pT181) immunolabel in brain homogenates as a proportion of total tau protein (Tau 5) in brain homogenates from mice exposed to single blast or sham blast 2 weeks before euthanizing. Mean values ± SEM in arbitrary densitometric units. P < 0.001, two-tailed Student’s t test.
Major question
Does blast exposure increase levels of hyperphosphorylated tau protein, the signature sign of CTE?
The authors observed microscopic evidence of blast-related tau pathology in the brain by immunohistochemistry (IHC). This figure shows the results of immunoblotting to both confirm and extend their observations.
Western blotting
The authors used Western blotting to measure changes in tau protein expression in blast-exposed mice. Western blotting is a common technique used to identify and quantify proteins in a sample.
First, samples are homogenized (mixed up) to break up the tissues and component cells. This 'homogenate' is mixed with a buffer and loaded onto a gel for gel electrophoresis. During gel electrophoresis, an electric current is applied that causes charged proteins to migrate through the gel. Depending on their size, proteins will migrate faster (smaller) or slower (larger). This is how protein separation is achieved.
Once proteins are separated, the target protein(s) are identified and quantified using specific antibodies. A stain is applied, and density of the stain can be used to calculate the amount of a specific protein in the sample.
The microscope uses an electron beam, passing it through the sample. Materials that are more electron dense will interact more and appear darker.
Results
The authors saw an increase in the amount of both phosphorylated and total tau protein in the brains of mice who had been exposed to blasts. This showed up as darker staining of tau protein bands on the gel.
They also observed changes in the distribution and location of tau protein bands on the gels. This suggest that blast exposure changes not only the amount of tau protein in the brain, but the expression pattern within the population of tau proteins expressed in the brain.
Tau protein naturally exists in multiple variations in the brain. Blast exposure appears to alter the amount of tau protein that is abnormally phosphorylated, as well as the distribution of the variants. The implications of the changes in distribution are unknown and currently being investigated.
Next questions
Given that the authors observed blast-induced damage in the brain at the light microscope and electron microscope level and the ability to quantify an increase in abnormal tau protein, the authors asked the question of whether the abnormalities they saw are associated with functional consequences (e.g., cognitive deficits) in the living mouse.
This question is the scientific equivalent of asking, "So what?"
Single-blast exposure persistently impairs axonal conduction and long-term potentiation of activity-dependent synaptic transmission in the hippocampus
We investigated the possibility that blast-related histopathological and ultrastructural abnormalities would be reflected in equally persistent functional impairments in hippocampal neurophysiology. Analysis of Schaffer collateral–evoked synaptic field potential input-output relations (fig. S23B) did not reveal an effect of blast exposure on baseline synaptic transmission at either 2 weeks or 1 month after blast exposure (fig. S24). However, axonal conduction velocity of CA1 pyramidal cell compound action potentials in the stratum alveus (fig. 23A) was significantly slowed 2 weeks after blast exposure, an effect that persisted for at least 1 month [Fig. 6, A and B; P < 0.05, repeated-measures multifactorial analysis of variance (ANOVA)].
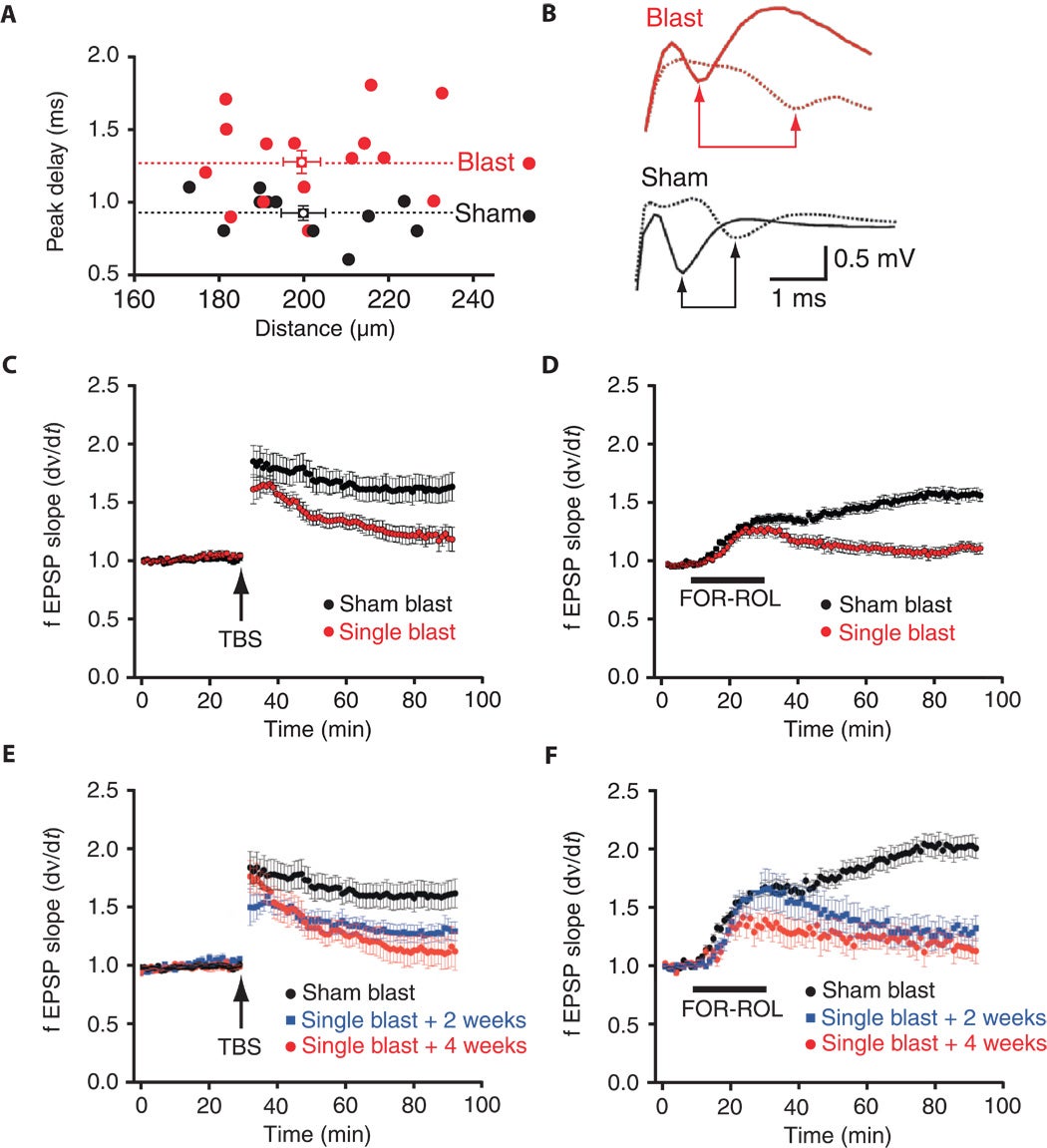
Fig. 6. Single-blast exposure induces persistent impairments in axonal conduction velocity and LTP of synaptic transmission in wild-type C57BL/6 mice. (A) Conduction velocity measurements of first peak compound action potential delay as a function of distance between recording electrodes in CA1 pyramidal cell axons in the stratum alveus of hippocampal slices from mice exposed to single blast (red circles, n = 13) compared to sham blast (black circles, n = 11). Mean ± SEM for each group. (B) Representative stimulus-evoked compound action potentials at proximal and distal recording sites (solid and hash lines, respectively) in hippocampal slices from mice exposed to single blast (red) and sham blast (black). Arrows indicate peak negativities used to calculate conduction velocity. (C) Time course of LTP at Schaffer collateral–CA1 synapses evoked by TBS in hippocampal slices from mice exposed to single blast (red circles, n = 17) compared to sham blast (black circles, n = 11). Each point mean ± SEM fEPSP slope of n slices. (D) Time course of LTP at Schaffer collateral–CA1 synapses evoked by bath application of the adenylate cyclase stimulant forskolin (50 μM) plus the type II phosphodiesterase inhibitor rolipram (10 μM; bar, FOR+ROL) in hippocampal slices from mice exposed to single blast (red circles, n = 27) compared to sham blast (black circles, n = 19). Each point mean ± SEM fEPSP slope of n slices. (E) Time course of LTP at Schaffer collateral–CA1 synapses evoked by TBS in hippocampal slices from mice 2 weeks (blue squares, n = 10) and 4 weeks after exposure to single blast (red circles, n = 7) compared to each other and to sham blast (black circles, n = 11). Each point mean ± SEM fEPSP slope of n slices. (F) Time course of long-lasting potentiation at Schaffer collateral–CA1 synapses evoked by bath application of the adenylate cyclase stimulant forskolin (50 μM) plus the type II phosphodiesterase inhibitor rolipram (10 μM; bar, FOR+ROL) in hippocampal slices from mice 2 weeks (squares, n = 12) and 4 weeks after exposure to single blast (red circles, n = 15) compared to each other and to sham blast (black circles, n = 19). Each point mean ± SEM fEPSP slope of n slices.
Major question
Using electrophysiological methods, the authors tested whether blast exposure disrupts two of the most important brain functions: neurotransmission and synaptic plasticity.
Neurotransmission is the transmission of electric signals between neurons. It is measured by axonal conduction (Panels A&B).
Synaptic plasticity is the ability of synapses to change, and is important to learning and memory. It is measured by long term potentiation (LTP) (Panels C-F).
Both of these functions were measured using hippocampal slice cultures.
See the experimental setup below:

Axonal conduction
Axonal conduction velocity can be measured with electrodes placed along an axon bundle.
Panel B shows axonal conduction in the hippocampus in a blast-exposed mouse (red) and a control mouse (black). The velocity is calculated by dividing the distance between the electrodes by the time it takes for the signal to travel between them. Panel A shows the delay in axonal conduction that results from blast exposure plotted against the distance between electrodes.
Results (axonal conduction)
As seen in panel B, the axonal conduction velocity in the hippocampus of blast-exposed mice is lower than in the hippocampus of sham control mice. This means it takes longer for the test signal ("tetanus") to travel from point A to point B.
Recall that the electron microscopy images showed axonal demyelination (i.e., damaged myelin sheaths). Myelin sheaths facilitate fast neurotransmission (i.e., nerve signal movement) down their length of the axon. The electrophysiology results here are consistent with electron microscopy observation showing axon demyelination.
Long-term potentiation (LTP)
Long term potentiation (LTP) is measured by conditioning a neuronal synapse with a high-frequency stimuli (using many successive electrical impulses or "tetanus"). Then, the effectiveness of transmitting a repeated test signal across the same pathway is measured.
The authors compared the rising slope of the propagated signal. They conditioned the pathway using theta-burst stimulation (TBS, Panels C and E). They then used a chemical treatment to train the pathway (Panels D and F).
Results (long term potentiation)
As the authors expected, the hippocampus in control mice showed a proper potentiated response, suggesting normal LTP synaptic plasticity. This can be seen in the elevated fEPSP slope in Panel C.
In the blast cases, the LTP effect is blocked. Almost immediately after spiking, the fEPSP slopes decrease to pre-training levels. Defective LTP was seen after two types of stimuli: electrical (TBS) and chemical (FOR-ROL). These different stimulation experiments can reveal important information abut the specific problems in the LTP pathway.
Next question
The authors have shown that neuron function in the brain is impaired by blast exposure. They next ask if these neurophysiological impairments lead to any cognitive or behavioral abnormalities in living mice.
Next, we examined the effect of blast exposure on stimulus- and cyclic adenosine monophosphate (cAMP)–evoked long-term potentiation (LTP) of synaptic strength at Schaffer collateral–CA1 synapses (fig. S23B), candidate mechanisms of memory storage. We found marked impairments of stimulus-evoked LTP in mouse slices prepared 2 weeks and 1 month after blast exposure (Fig. 6C; P < 0.05, repeated-measures multifactorial ANOVA). When the 2-week and 1-month blast-exposed cohorts were examined independently, we found that the magnitude of posttetanic potentiation (PTP) immediately after application of theta-burst stimulation (TBS) was significantly less at the 2-week time point (Fig. 6E; P < 0.05, repeated-measures multifactorial ANOVA). Although PTP recovered by 1 month after blast, the magnitude of LTP 1 hour after tetanus was significantly reduced at both postblast time points (Fig. 6E; P < 0.05, repeated-measures multifactorial ANOVA). These results indicate that exposure to single blast impaired long-term activity-dependent synaptic plasticity for at least 1 month after blast exposure in our model. Next, we examined cAMP-dependent LTP of Schaffer collateral–CA1 field excitatory postsynaptic potentials (fEPSPs) induced by 15-min bath application of the adenylate cyclase activator forskolin (50 μM) plus the type II phosphodiesterase inhibitor rolipram (10 μM). In contrast to control slices, cAMP-LTP was profoundly attenuated 30 to 60 min after drug washout in hippocampal slices prepared from both left and right hemispheres of mice 2 weeks and 1 month after blast exposure (Fig. 6D and fig. S25, A and B; P < 0.05, repeated-measures multifactorial ANOVA). As with stimulus-evoked LTP, cAMP-LTP was equally impaired at both 2 weeks and 1 month after blast exposure, demonstrating the long-term nature of blast effects on both activity-dependent and chemically evoked synaptic plasticity (Fig. 6F; P < 0.05, repeated-measures multifactorial ANOVA).
Single-blast exposure induces long-term behavioral deficits that are prevented by head immobilization during blast exposure
We did not detect significant differences between single-blast and sham-blast mice in total distance, mean velocity, or central zone entries in open-field behavior testing (Fig. 7, A to C), indicating that blast exposure did not impair gross neurological functioning with respect to locomotion, exploratory activity, and thigmotaxis (an indicator of murine anxiety assessed by movement close to the wall of the experimental apparatus). In contrast, when we tested acquisition and long-term retention of hippocampal-dependent spatial learning and memory in the Barnes maze (Fig. 7, D to F), we observed that blast-exposed mice exhibited significantly longer escape latencies (Fig. 7D; P < 0.05, two-way ANOVA) and poorer memory retrieval 24 hours after the final training session (Fig. 7E; P < 0.05, Student’s t test) compared to sham-blast control mice. These findings are consistent with persistent blast-related hippocampal dysfunction.
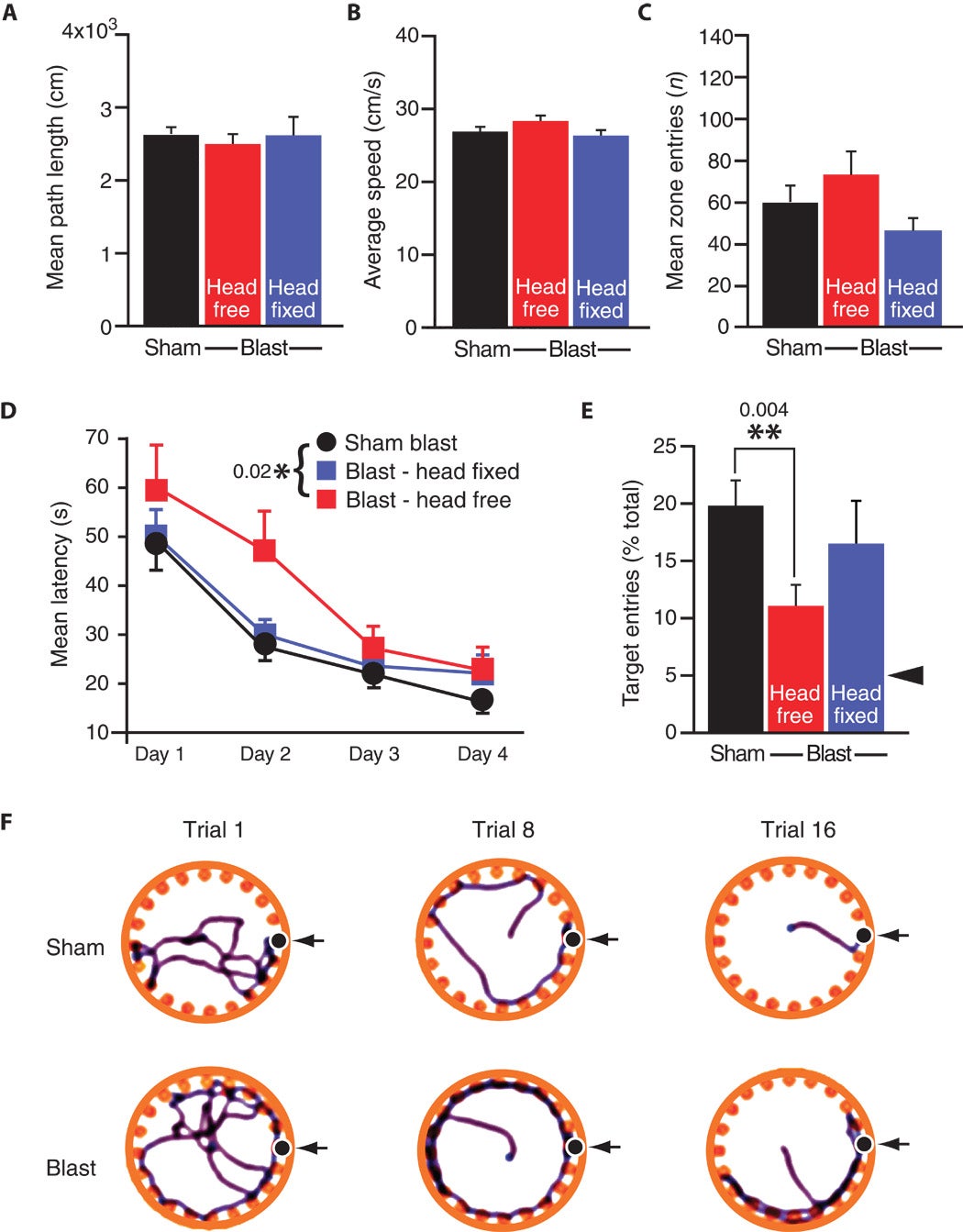
Fig. 7. Single-blast exposure in wild-type C57BL/6 mice induces persistent hippocampal-dependent learning and memory deficits that are prevented by head fixation (immobilization) during blast exposure. (A to C) Open-field testing showed no effect of blast exposure on gross locomotor function, explorative activity, or thigmotaxis as measured by total distance traveled (A), mean velocity (B), and number of central zone entries (C), respectively, in mice exposed to single blast (red bars, single blast, head free, n = 10; blue bars, single blast, head fixed, n = 10) or sham blast (black bars, sham blast, n = 20). (D to F) Barnes maze testing demonstrated significant impairments in hippocampal-dependent spatial learning acquisition measured by decreasing latency to find the escape box across 4 days of training (D) (two-way ANOVA, P = 0.020) and long-term memory assessed by escape box location recall assessed 24 hours after the last training session (E) (**P = 0.004, Student’s t test). Mice exposed to single blast (red squares, single blast, head free, n = 10) are compared to pooled sham-blast control mice (circles, sham blast, n = 20). Fixation (immobilization) of the head during blast exposure (blue squares, single blast, head fixed, n = 10) reversed blast-induced learning and memory deficits. Arrowhead in (E) represents 5% level predicted by chance selection of the escape box from among the 20-hole choices. (F) Representative Barnes maze tracks obtained on trials 1, 8, and 16 for mice exposed to a single blast (bottom row) compared to sham blast (top row).
Major question
In Figure 6, the authors show that there are functional impairments in the hippocampus of blast-exposed mice. The hippocampus is critical to learning and memory formation.
In this figure, they address the question of whether blast-related impairments interfere with contextual learning and memory retention. They also ask if removing the hypothesized injury mechanism (traumatic head motion during blast exposure) would reduce or prevent blast-related learning and memory deficits.
Barnes maze
The Barnes maze is a well-established test of spatial learning and memory retention. Panel F shows an illustration of the Barnes maze setup. It is a circular table with holes around the perimeter of the round platform. Only one hole leads to an escape box.
When mice are placed in the center, a normal mouse will will explore the environment and eventually find the exit. On repeated testing, the same mouse will take less time to find the escape hole. This decrease in time (latency) is a direct measure of spatial learning.
Mouse activity during the test can be tracked with a video camera that has specialized software which quantifies the behavioral analysis.
Locomotor function (Panels A-C)
The authors first showed that blast-exposed mice and control mice have normal locomotor function (movement ability) during the Barnes maze. The authors did not see significant differences between the groups.
Thus, any differences in Barnes maze testing could not be because of differences in locomotor activity.
Results
Blast-exposed mice showed significant impairments in spatial learning. This was evidenced by the fact that they took longer to find the escape hole when they were repeatedly put into the maze.
The authors also saw that blast-exposed mice had impaired memory retention for the same task. Deficits in spatial learning and memory retention are both consistent with defective LTP in the hippocampus. They are also common cognitive deficits seen in blast-exposed military veterans.
Panel F shows two example cases:
- In trial 1, the sham (control) mouse explores the table for an exit. By trial 8, the same mouse takes less time to find the escape hole. By trial 16, the mouse moves quickly to the exit.
- In trial 1, the blast-exposed mouse takes much more time to locate the exit. It shows greater difficulty in remembering this spatial information, which is evidenced by the fact that in trials 8 and 16 it wanders around more than the control example.
The authors also observed that restraining the head during blast exposure prevents the cognitive deficits observed in mice whose heads were allowed to move freely.This confirms the authors' hypothesis that traumatic head motion contributes to the cognitive deficits associated with blast exposure (the "bobblehead effect").
The results of kinematic analysis (Fig. 2, D to G) suggested that blast-induced head acceleration was a likely pathogenic mechanism by which blast exposure leads to TBI and neurobehavioral sequelae. To test this hypothesis, we compared hippocampal-dependent learning acquisition and memory retention in mice with and without head immobilization during single-blast exposure and in sham-blast control mice. Head immobilization during blast exposure eliminated blast-related impairments in hippocampal-dependent learning acquisition (Fig. 7D; P > 0.20, repeated-measures ANOVA with post hoc Scheffe test compared to sham-blast controls) and restored blast-related memory retention deficits to normal levels (Fig. 7E; P > 0.20, one-way ANOVA with post hoc Scheffe test), supporting the conclusion that head acceleration is necessary for behavioral learning impairments.
DISCUSSION
We analyzed a case series of postmortem human brains from U.S. military veterans with blast exposure and/or concussive injury and compared them to brains from young-adult athletes with histories of concussive injury and from normal controls of comparable ages without histories of blast exposure, concussive injury, or neurological disease. We uncovered evidence of CTE-linked tau neuropathology, including multifocal perivascular foci of neurofibrillary and glial tangles immunoreactive for phosphorylation-independent (Tau-46) and phosphorylation-dependent (CP-13) tau epitopes (20, 21), in the brains of blast-exposed and/or concussive-injured veterans. This blast-associated CTE-linked tau neuropathology was indistinguishable from the tau neuropathology, neuroinflammation, and neurodegeneration observed in the brains of young-adult athletes with histories of repeat concussive injury. Examination of brains from wild-type C57BL/6 mice 2 weeks after exposure to a single controlled blast also revealed histopathological, ultrastructural, and biochemical evidence of CTE-linked neuropathology, including tau protein–linked immunoreactivity, persistent perivascular pathology, cortical and hippocampal neurodegeneration, myelinated axonopathy, chronic neuroinflammation with widespread astrocytosis and microgliosis, and phosphorylated tau proteinopathy. Overall, our findings of persistent CTE-linked neuropathology in the brains of military veterans with blast exposure and/or concussive injury and young athletes with repeat concussive injury suggest that TBI induced by different insults under different conditions can trigger common pathogenic mechanisms leading to similar neuropathology and sequelae. Notably, within this small controlled case series, the effects of blast exposure, concussive injury, and mixed trauma (blast exposure and concussive injury) were indistinguishable.
Experimental results from our murine blast neurotrauma model provide evidence linking blast exposure with development of CTE-like tau neuropathology. Moreover, this blast-related neuropathology was associated with persistent neurophysiological and cognitive deficits that recapitulate clinical signs and symptoms reported in military veterans with blast-related TBI (2, 7, 9–15) and concussive-injured athletes diagnosed with CTE (20, 21). It is notable that exposure to a single blast in our mouse model was sufficient to induce early CTE-like neuropathology, slowed axonal conduction velocity, and defective stimulus- and cAMP-dependent LTP of synaptic transmission. Notably, these blast-related neurophysiological abnormalities were contemporaneous with somatodendritic alterations in hippocampal and cortical total tau and phosphorylated tau neuropathology and biochemistry, microvascular ultrastructural pathology, and impairment in hippocampal-dependent learning acquisition and memory retention.
Although blast-exposed C57BL/6 mice recapitulated key features of human CTE neuropathology, including cellular accumulation of phosphorylated tau protein and pre-tangle tau protein neuropathology, it is notable that mature NFTs were not detected in the cortex or hippocampus of blast-exposed mice. This apparent discordance with human CTE neuropathology may be explained by the early time points chosen for evaluation in our mouse studies or, alternatively, as a forme fruste resulting from resistance of wild-type murine tau protein to form neurotoxic aggregates in vivo. However, our results demonstrate blast-related immunohistochemical and biochemical abnormalities in tau hyperphosphorylation at the 2-week time point after single-blast exposure. Studies of triple-transgenic mice expressing human tau protein and human amyloid-β peptide have shown that controlled cortical impact injury leads to rapid accumulation of hyperphosphorylated tau within 24 hours after experimental injury (49, 50). These findings suggest that genotypic determinants may be critical factors that modulate temporal and phenotypic expression of TBI and late-emerging sequelae, including CTE.
ICP dynamics recorded during blast exposure revealed blast-induced pressure transients in the hippocampus that were coincident with and comparable in amplitude, waveform, and impulse to FFP measurements outside the cranium. This finding is consistent with the head acting as a lumped element for which the blast-induced external pressure differential equilibrates within ~100 μs. Measured blast pressure amplitudes in the brain were on the order of 100 kPa (~1 bar), a magnitude equivalent to water pressure at a depth of ~10 m. Although it is possible that high-frequency components (>100 kHz) could lead to localized focusing due to reverberation and constructive interference, the pressure amplitudes we measured were far below tissue damage thresholds. Tissue damage associated with clinical ultrasound requires negative acoustic pressures in excess of 1 MPa (51) that lead to excitation of cavitation bubbles. Thresholds for positive pressures are not well characterized but are likely to exceed 40 MPa because positive pressures commonly used in clinical shock wave lithotripsy are not associated with significant, if any, tissue damage (52). Thresholds for tissue damage from underwater sonar require ~100 kPa (53) and result from many cycles of bubble growth and collapse over tens of seconds of continuous wave excitation. Tissue damage in this setting is due to the negative pressure rather than exposure to a single compression pulse. These considerations indicate that direct tissue damage resulting from transmission of the blast shock wave through the brain is unlikely. Our results indicate that ICP transients closely approximate FFP measurements in air (27, 54, 55). Moreover, blast wavefront transmission was identical when measured in the brain of intact living mice or isolated mouse heads severed at the cervical spine, suggesting that neither thoracic-mediated mechanisms nor vascular hemodynamic effects contributed significantly to ICP transients during blast exposure. Together, our findings point to the substantial inertial forces and oscillating acceleration-deceleration cycles imposed on the head by blast wind (bobblehead effect) as the primary biomechanical mechanism by which blast exposure initiates acute closed-head brain injury and sequelae, including CTE (fig. S26).
Here, we describe CTE-linked neuropathology in the brains of military veterans with blast exposure and/or concussive injury, young-adult athletes with repetitive concussive injury, and mice subjected to a single blast. These observations are consistent with a common injury mechanism involving oscillating head acceleration-deceleration cycles (bobblehead effect; fig. S26) that lead to pathogenic shearing strain imposed on the cranial contents (56–60). Our observation that head immobilization during blast exposure prevented hippocampal-dependent learning and memory deficits in blast-exposed mice provides additional support for this injury mechanism and postulated relationship to persistent neurobehavioral sequelae. Recent studies have identified local strain amplification near micromechanical heterogeneities in the brain, including sulci, blood vessels, and axons as possible contributory factors leading to blast-related brain injury (48, 61, 62). Simulation studies indicate that pressure gradients in the brain of an unhelmeted head resulting from military blast exposure may be sufficiently large to generate damaging intracranial forces, even in the absence of direct impact trauma to the head (63). Ultrastructural analysis indicates that blast exposure in our experimental model was associated with persistent microvascular pathology, including abnormal blood-brain barrier (BBB) cytoarchitecture. Blast-related ultrastructural pathology may be associated with pericyte degeneration (64) and/or microvascular compression secondary to astrocytic end-feet swelling (65), thereby leading to BBB compromise, local hypoxia, chronic neuroinflammation, and neurodegeneration. These postulated mechanisms are consistent with recent experimental studies (66–68), finite element head modeling analyses (60, 69), and the results in this report. Further studies are needed to differentiate the relative contributions of these and possibly other pathogenic mechanisms.
The significance of the neurophysiological abnormalities in blast-exposed wild-type C57BL/6 mice is substantial. First, although blast exposure did not produce detectable long-term dysfunction in basal synaptic transmission, exposure to a single sublethal blast was sufficient to induce profound and persistent impairment of both activity- and cAMP-dependent LTP in hippocampal CA1 pyramidal neurons, candidate cellular mechanisms of long-term memory processing. The fact that both forms of LTP require dendritic protein synthesis (70, 71) and gene transcription (72) indicate that blast exposure may induce long-lasting damage to cellular signal transduction downstream of synaptic glutamate release. Mechanisms that may be altered by blast exposure include N-methyl-D-aspartate glutamate receptor activation, intracellular second messenger systems, gene expression, protein synthesis, and posttranslational modification. Our results also indicate that blast exposure can induce persistent axonal conduction defects that further impair cognitive processing and are consistent with recent findings from human studies (14, 15). These effects may be mediated by diffuse axonal injury, Wallerian degeneration, and/or differential susceptibility of larger neurons to structural or functional axotomy. Damage to these and other brain structures, systems, and mechanisms may contribute to abnormalities in neurochemical homeostasis, cerebral metabolism, and neurophysiological functions associated with blast-related TBI (73). Our results suggest that blast exposure may hold comparable or even greater pathogenic potential than repetitive head injury associated with contact athletics (74–76).
Limitations of the human neuropathology reported here include the small number of available cases, the time interval between trauma and postmortem examination, potential contributions of confounding comorbidities and risk factors, and inherent limitations of neuropathological analysis to establish mechanistic causality. Clinicopathological correlation may be further complicated by genetic contributions [for example, APOE (apolipoprotein E) genotype (77)], history of previous head trauma, innate inflammatory responsivity, neuropsychiatric comorbidity, age and gender, and other factors with potential to modulate susceptibility and pathological expression of blast-related neurotrauma and sequelae. Furthermore, emerging evidence indicates that PTSD may represent an important overlapping comorbidity with potential to synergistically affect both the incidence and the severity of blast TBI and military deployment–related cognitive dysfunction (2, 7, 78–80). Limitations of our animal experiments include use of adult male C57BL/6 wild-type mice subjected to a single-blast exposure with post-exposure evaluation at time points only up to 1 month. Interpretation and generalizability of our animal experiments are further constrained by interspecies differences, including the significantly greater deformability of the murine skull, the relative instability of the murine cervical spine, and differential force loading on the head and neck in mice and humans.
Our results provide compelling evidence linking blast exposure to long-lasting brain injury. Specifically, our study raises concern that blast exposure may increase risk for later development of CTE and associated neurobehavioral sequelae. Indeed, the severity, persistence, and possible progression of the neuropathological abnormalities and neurophysiological deficits observed in our study indicate that blast exposure is a potent insult with enduring pathogenic potential and functional significance. The availability of a neuropathologically validated murine model with correspondence to human CTE is expected to open new avenues for investigation of mechanisms, biomarkers, and risk factors relevant to blast-related brain injury and facilitate development of urgently needed diagnostics, therapeutics, and prophylactic measures for blast neurotrauma and its aftermath.
MATERIALS AND METHODS
Human subjects
The brain and spinal cord of 12 human subjects (male military veterans, ages 22 to 45 years, mean 32.3 years, with histories of explosive blast and/or concussive injury 1 to 6 years before death, n = 4; male athletes with histories of repetitive concussive injury, including 3 amateur American football players and a professional wrestler, ages 17 to 27 years, mean 20.8 years, n = 4; male normal controls, ages 18 to 24 years, mean 20.5 years, without known blast exposure, trauma history, or neurological disease, n = 4) were procured through the Boston University Alzheimer’s Disease Center and Center for the Study of Traumatic Encephalopathy at Boston University School of Medicine. Blast exposure, trauma history, and neurological status at the time of death were determined through review of medical records and interviews with next of kin. Ethical permission to conduct this investigation was approved by Institutional Review Board at Boston University School of Medicine. The study conforms to institutional regulatory guidelines and principles of human subject protection in the Declaration of Helsinki.
Animal subjects
Adult wild-type C57BL/6 male mice (Charles River Laboratories) were group-housed at the Laboratory Animal Science Center, Boston University School of Medicine. All animal experiments used 2.5-month-old mice with 8 to 10 mice per group. Animal housing and experimental use were conducted in accordance with Association for Assessment and Accreditation of Laboratory Animal Care guidelines, in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals, and adherence to principles in the National Research Council Guide for the Care and Use of Laboratory Animals. All studies were approved by Institutional Animal Care and Use Committees at Boston University School of Medicine and New York Medical College.
Histopathological and electron microscopic analyses
Postmortem human brain and spinal cord were received as fresh tissue and as fixed tissue in formalin after processing by medical examiners. Neuropathological analysis followed established protocols at the Boston University Alzheimer’s Disease Center and included comprehensive examination for all neurodegenerative conditions (81). Paraffin-embedded sections from at least 15 brain regions were stained with Luxol fast blue, hematoxylin and eosin, and Bielschowsky silver stain. Mice were euthanized by CO2 asphyxiation and transcardially perfused with phosphate-buffered saline (PBS). Whole brains were prefixed in 10% neutral buffered formalin, block-sectioned into 2-mm coronal slabs, postfixed in 4% paraformaldehyde, paraffin-embedded, and serially sectioned at 10 μm. A battery of primary detection antibodies (table S1) was used for immunohistopathological analyses. Ultrastructural studies were conducted on fixed brain specimens embedded in Epon, sectioned at 60 nm, stained with uranyl acetate or lead citrate, and examined with a Tecnai-G2 Spirit BioTWIN electron microscope with an AMT 2K CCD camera.
Murine blast neurotrauma model system
A compressed gas-driven shock tube (fig. S3) was developed in collaboration with the Fraunhofer Center for Manufacturing Innovation at Boston University (Brookline, MA) and installed at the Neurotrauma Laboratory, Boston University School of Medicine. This instrument was used to deliver highly reproducible blast waves (Fig. 2A, figs. S3 to S7, and tables S2 and S3). Adult wild-type C57BL/6 male mice (2.5 months) were anesthetized with ketamine (75 mg/kg, intraperitoneally), xylazine (4.3 mg/kg, intraperitoneally), and buprenorphine (0.2 mg/kg, subcutaneously) and secured in the prone position in a thoracic-protective restraint system inside the shock tube (fig. S3). The head and neck were free to allow flexion, extension, and rotation of the cervical spine in the horizontal and sagittal planes of motion to model conditions relevant to military blast exposure. Maximum burst pressure compatible with 100% survival and no gross motor abnormalities was ascertained empirically (table S2). Experimental blast parameters (incident static pressure, 77 ± 2 kPag; blast overpressure rise time, 38 ± 3 μs; compressive phase duration, 4.8 ± 0.1 ms; shock wave velocity, 1.26 ± 0.04 Mach; calculated blast wind velocity, 150 m/s = 336 miles/hour; table S2) closely approximate explosive blast produced by detonation of 5.8 kg of TNT measured at a standoff distance of 5.5 m [ConWep analysis (44); table S3]. This blast exposure is within the range of typical IED detonations and standoff distances associated with military blast injury (45). Anesthetized mice were exposed to a single blast or sham blast, removed from the apparatus, monitored until recovery of gross locomotor function, and then transferred to their home cage.
Static and reflected FFP measurements
Assessment of static and reflected FFP was assessed by two piezoelectric pressure sensors (model HM102A15, PCB Piezotronics) placed in the shock tube at the same axial distance relative to the head of the animal subjects. A static pressure (side-on) sensor was flushed-mounted inside the shock tube. A second transducer was positioned with the detector facing into the shock tube in a reflected pressure (face-on) orientation. Pressure signals were processed with a PCB signal conditioner (model 482C05, PCB Piezotronics) and recorded at a frequency of 5 MHz with a digital oscilloscope (640Zi WaveRunner, LeCroy). Voltages were converted to pressure with calibration data provided by the manufacturer and processed with 2-kHz low-pass filtering.
ICP measurements
ICP measurements were conducted with a broad-bandwidth piezoelectric needle hydrophone (NP10-3, DAPCO Industries) with a 0.6-mm-diameter element sheathed in a stainless steel hypodermic needle. Pressure sensitivity was flat to within ±3 dB for frequencies ranging from 1 Hz to 170 kHz. The needle hydrophone was inserted into the hippocampus at −3.00 mm caudal to the bregma suture, +3.50 mm lateral to the sagittal suture, and +2.00 mm ventral to the skull surface. For ICP measurements, the head was immobilized to prevent displacement of the pressure sensor. Piezoelectric voltage signals were recorded by a digital oscilloscope (640Zi WaveRunner, LeCroy) and converted to pressure units with calibration data supplied by the manufacturer and processed with 20-kHz low-pass filtering. Post-acquisition processing was performed with Matlab 2009 (MathWorks).
High-speed videographic kinematic analysis
High-speed videography was conducted with a FASTCAM SA5 camera (Photron USA Inc.; courtesy of Tech Imaging) operated at 10-μs frame capture rate. Videographic records were reassembled with open-source ImageJ software and processed in Matlab (MathWorks). Angular position and motion of the head were assessed by tracking a reflective paint mark on the snout, calculated by assuming a central pivot point between the scapulae (fig. S3B), and processed with 500-Hz low-pass filtering (Fig. 2, D to G, fig. S3B, and video S1).
Hippocampal electrophysiology
Mice were decapitated under deep isoflurane anesthesia, and the brains were quickly removed, hemisected, and sectioned with a Leica model VT 1200S vibratome at 350 μm. Slices were fixed to a stage with cyanoacrylate adhesive and immersed in oxygenated artificial cerebrospinal fluid (126 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 1.3 mM MgCl2, 2.5 mM CaCl2, 26 mM NaHCO3, 10 mM glucose, saturated with 95% O2 and 5% CO2) at 32°C. Experimental drugs were bath applied in the perfusate at a rate of 3 ml/min. Axonal conduction velocity was assessed with a recording electrode placed in CA1 stratum alveus. Schaffer collateral–CA1 synaptic transmission and plasticity were assessed with a recording electrode in the CA1 stratum radiatum.
Hippocampal-dependent learning and memory
Open-field testing (Med-Associates) was used to assess gross locomotor function, exploratory activity, and thigmotaxis. Hippocampal-dependent learning acquisition and memory retention were evaluated in the Barnes maze (82). Spatial learning was assisted by visual cues in the environment that remained constant across test sessions. Movement was tracked and recorded electronically (Stoelting). Latency to find the escape box, trajectory velocity to the escape box, and total trajectory distance were assessed and recorded daily in four sessions conducted over 4 days. Memory retrieval was electronically assessed by recording the number of nose pokes in blank holes as a percentage of total nose pokes recorded 24 hours after completion of the learning protocol.
Quantitative assessment of phosphorylated and total tau protein
Quantitative immunoblot analysis was conducted with left and right hemisected brains obtained from PBS-perfused mice 2 weeks after exposure to a single blast (n = 6 mice) or sham blast (n = 6 mice). Snap-frozen hemisected brain specimens were thawed, resuspended in 0.7 ml of protease-phosphatase inhibitor buffer, and homogenized as previously described (83). Protein concentrations were normalized and equal sample volumes were subjected to standard polyacrylamide gel electrophoresis in duplicate. Immunoblot detection used monoclonal antibody AT270 (Innogenetics) directed against tau protein phosphorylated at Thr181 (pT181), monoclonal antibody CP-13 (P. Davies, Albert Einstein College of Medicine) directed against tau protein phosphorylated at Ser202 (pS202) and Thr205, or monoclonal antibody Tau 5 (L. Binder, Northwestern University Medical School) directed against phosphorylation-independent tau protein. Triplicate densitometry measurements were analyzed with open-source ImageJ software. A commercial ELISA kit was used to quantitate murine-specific tau protein phosphorylated at Ser199 (Invitrogen). Frozen brain samples were homogenized in eight volumes of 5 M guanidine-HCl and 50 mM tris (pH 8) followed by five passes in a glass Teflon homogenizer. Homogenates were mixed for 3 hours, diluted into PBS containing protease inhibitors, and centrifuged for 20 min at 16,000g. Supernatants were diluted and assayed in quadruplicate according to the manufacturer’s instructions.
Statistical analyses
Comparisons of axonal conduction velocity and LTP magnitude were conducted with repeated-measures multifactorial ANOVA with Bonferroni-Dunn post hoc correction. Longitudinal neurobehavioral data were analyzed by repeated-measures ANOVA. Memory retrieval was evaluated by ANOVA. Statistical significance was preset at P < 0.05.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/4/134/134ra60/DC1
Materials and Methods
References
Table S1. Summary of antibodies used in this study.
Table S2. Murine blast neurotrauma model blast parameters.
Table S3. Shock tube blast compared to equivalent explosive blast.
Fig. S1. Phosphorylated tau axonopathy in a single axon from the brain of a 22-year-old male military veteran with exposure to a single improvised explosive device blast and persistent blast-related traumatic brain injury symptoms.
Fig. S2. Absence of CTE neuropathology in a representative postmortem human brain from 21-year-old male control subject without known history of blast exposure or concussive injury.
Fig. S3. Schematic and geometry of the murine blast neurotrauma shock tube system.
Fig. S4. Reproducibility of shock tube blast static and reflected pressure.
Fig. S5. Peak reflected and static incident pressure as a function of shock tube burst pressure.
Fig. S6. Shock wave velocity (Mach) regression analysis.
Fig. S7. X-T wave diagram demonstrating positional and temporal features of the blast shock wave.
Fig. S8. Unperfused C57BL/6 mouse brain 2 weeks after single shock tube blast exposure.
Fig. S9. Neuropathology in the CA3 field and dentate gyrus in a C57BL/6 mouse brain 2 weeks after exposure to a single shock tube blast.
Fig. S10. Decreased choline acetyltransferase (ChAT) immunoreactivity in the brainstem and neuronal dropout in the cerebellum of C57BL/6 mice 2 weeks after exposure to a single shock tube blast.
Fig. S11. Electron micrographic montage of the hippocampus CA1 field in a C57BL/6 mouse brain 2 weeks after exposure to a single shock tube blast.
Fig. S12. High-magnification electron micrographs of the hippocampus CA1 field in a C57BL/6 mouse brain 2 weeks after single-blast exposure.
Fig. S13. Perivascular ultrastructural pathology in the hippocampus CA1 stratum radiatum in a C57BL/6 mouse brain 2 weeks after exposure to a single shock tube blast.
Fig. S14. Perivascular ultrastructural pathology in the hippocampus CA1 stratum radiatum in a C57BL/6 mouse brain 2 weeks after exposure to a single shock tube blast.
Fig. S15. Perivascular ultrastructural pathology in the hippocampus CA1 stratum radiatum in a C57BL/6 mouse brain 2 weeks after exposure to a single shock tube blast.
Fig. S16. Perivascular ultrastructural pathology in the hippocampus CA1 stratum radiatum in a C57BL/6 mouse brain 2 weeks after exposure to a single shock tube blast.
Fig. S17. Myelin figure in the hippocampus CA1 stratum pyramidale in a C57BL/6 mouse brain 2 weeks after exposure to a single shock tube blast.
Fig. S18. A microglial cell amidst myelinated axons in the hippocampus CA1 stratum alveus in a C57BL/6 mouse brain 2 weeks after exposure to a single shock tube blast.
Fig. S19. Autophagy and mitophagy in the hippocampus CA1 field in a C57BL/6 mouse brain 2 weeks after exposure to a single shock tube blast.
Fig. S20. Degenerating (“dark”) pyramidal neurons in the hippocampus CA1 stratum pyramidale in a C57BL/6 mouse brain 2 weeks after exposure to a single shock tube blast.
Fig. S21. Degenerating (dark) pyramidal neurons in the hippocampus CA1 stratum pyramidale in a C57BL/6 mouse brain 2 weeks after exposure to a single shock tube blast.
Fig. S22. Degenerating (dark) pyramidal neurons in the hippocampus CA1 stratum pyramidale in a C57BL/6 mouse brain 2 weeks after exposure to a single shock tube blast.
Fig. S23. Electrode placements for axonal conduction velocity and synaptic plasticity experiments.
Fig. S24. Schaffer collateral–CA1 synaptic input-output relations illustrating the absence of long-term effects of blast exposure on baseline synaptic transmission.
Fig. S25. Blast-induced deficits in cAMP-induced long-term potentiation of synaptic transmission at Schaffer collateral–CA1 synapses are bilateral and persistent.
Fig. S26. Model of blast- and concussion-related TBI and sequelae, including CTE.
Video S1. Mouse head kinematics during exposure to a single shock tube blast.
REFERENCES AND NOTES
- TRITON Report (Allen-Vanguard Threat Solutions, Shrivenham, UK, 2011).
- C. W. Hoge, D. McGurk, J. L. Thomas, A. L. Cox, C. C. Engel, C. A. Castro, Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N. Engl. J. Med. 358, 453–463 (2008).
- S. J. Wolf, V. S. Bebarta, C. J. Bonnett, P. T. Pons, S. V. Cantrill, Blast injuries. Lancet 374, 405–415 (2009).
- Independent Panel on the Safety and Security of United Nations Personnel in Iraq, http://www.un.org/News/dh/iraq/safety-security-un-personnel-iraq.pdf [accessed 24 February 2012].
- K. H. Taber, D. L. Warden, R. A. Hurley, Blast-related traumatic brain injury: What is known? J. Neuropsychiatry Clin. Neurosci. 18, 141–145 (2006).
- C. K. Murray, J. C. Reynolds, J. M. Schroeder, M. B. Harrison, O. M. Evans, D. R. Hospenthal, Spectrum of care provided at an echelon II Medical Unit during Operation Iraqi Freedom. Mil. Med. 170, 516–520 (2005).
- T. L. Tanielian, L. H. Jaycox, Invisible Wounds of War: Psychological and Cognitive Injuries, Their Consequences, and Services to Assist Recovery (RAND Corporation, Santa Monica, CA, 2008).
- H. Terrio, L. A. Brenner, B. J. Ivins, J. M. Cho, K. Helmick, K. Schwab, K. Scally, R. Bretthauer, D. Warden, Traumatic brain injury screening: Preliminary findings in a US Army Brigade Combat Team. J. Head Trauma Rehabil. 24, 14–23 (2009).
- G. Ling, F. Bandak, R. Armonda, G. Grant, J. Ecklund, Explosive blast neurotrauma. J. Neurotrauma 26, 815–825 (2009).
- T. L. Benzinger, D. Brody, S. Cardin, K. C. Curley, M. A. Mintun, S. K. Mun, K. H. Wong, J. R. Wrathall, Blast-related brain injury: Imaging for clinical and research applications: Report of the 2008 St. Louis workshop. J. Neurotrauma 26, 2127–2144 (2009).
- J. J. Vasterling, M. Verfaellie, K. D. Sullivan, Mild traumatic brain injury and posttraumatic stress disorder in returning veterans: Perspectives from cognitive neuroscience. Clin. Psychol. Rev. 29, 674–684 (2009).
- R. R. Hicks, S. J. Fertig, R. E. Desrocher, W. J. Koroshetz, J. J. Pancrazio, Neurological effects of blast injury. J. Trauma 68, 1257–1263 (2010).
- L. A. Brenner, B. J. Ivins, K. Schwab, D. Warden, L. A. Nelson, M. Jaffee, H. Terrio, Traumatic brain injury, posttraumatic stress disorder, and postconcussive symptom reporting among troops returning from Iraq. J. Head Trauma Rehabil. 25, 307–312 (2010).
- E. R. Peskind, E. C. Petrie, D. J. Cross, K. Pagulayan, K. McCraw, D. Hoff, K. Hart, C. E. Yu, M. A. Raskind, D. G. Cook, S. Minoshima, Cerebrocerebellar hypometabolism associated with repetitive blast exposure mild traumatic brain injury in 12 Iraq war veter-ans with persistent post-concussive symptoms. Neuroimage 54 (Suppl. 1), S76–S82 (2011).
- C. L. Mac Donald, A. M. Johnson, D. Cooper, E. C. Nelson, N. J. Werner, J. S. Shimony, A. Z. Snyder, M. E. Raichle, J. R. Witherow, R. Fang, S. F. Flaherty, D. L. Brody, Detection of blast-related traumatic brain injury in U.S. military personnel. N. Engl. J. Med. 364, 2091–2100 (2011).
- Department of Veterans Affairs and Department of Defense, Clinical practice guideline: Management of concussion/mild traumatic brain injury; http://www.healthquality.va.gov/management_of_concussion_mtbi.asp [accessed 7 March 2012].
- D. K. Menon, K. Schwab, D. W. Wright, A. I. Maas; Demographics and Clinical Assessment Working Group of the International and Interagency Initiative toward Common Data Elements for Research on Traumatic Brain Injury and Psychological Health, Positionstatement: Definition of traumatic brain injury. Arch. Phys. Med. Rehabil. 91, 1637–1640 (2010).
- H. Miller, Mental after-effects of head injury. Proc. R. Soc. Med. 59, 257–261 (1966).
- B. I. Omalu, S. T. DeKosky, R. L. Minster, M. I. Kamboh, R. L. Hamilton, C. H. Wecht, Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 57, 128–134 (2005).
- A. C. McKee, R. C. Cantu, C. J. Nowinski, E. T. Hedley-Whyte, B. E. Gavett, A. E. Budson, V. E. Santini, H. S. Lee, C. A. Kubilus, R. A. Stern, Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 68, 709–735 (2009).
- A. C. McKee, B. E. Gavett, R. A. Stern, C. J. Nowinski, R. C. Cantu, N.W. Kowall, D. P. Perl, E. T. Hedley-Whyte, B. Price, C. Sullivan, P. Morin, H. S. Lee, C. A. Kubilus, D. H. Daneshvar, M. Wulff, A. E. Budson, TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J. Neuropathol. Exp. Neurol. 69, 918–929 (2010).
- H. Celander, C. J. Clemedson, U. A. Ericsson, H. I. Hultman, The use of a compressed air operated shock tube for physiological blast research. Acta Physiol. Scand. 33, 6–13 (1955).
- D. R. Richmond, E. G. Damon, I. G. Bowen, E. R. Fletcher, C. S. White, Air-blast studies with eight species of mammals. Techn Progr Rep DASA 1854. Fission Prod. Inhal. Proj. 1–44 (1967).
- Y. Phillips, D. Richmond, in Textbook of Military Medicine. Part I. Warfare, Weapons, and the Casualty, R. Zatchuck, D. Jenkins, R. Bellamy, C. Quick, Eds. (TMM Publications, Washington, DC, 1990), vol. 5, pp. 221–240.
- C. Kaur, J. Singh, M. K. Lim, B. L. Ng, E. P. Yap, E. A. Ling, The response of neurons and microglia to blast injury in the rat brain. Neuropathol. Appl. Neurobiol. 21, 369–377 (1995).
- N. M. Elsayed, Toxicology of blast overpressure. Toxicology 121, 1–15 (1997).
- A. Säljö, F. Arrhén, H. Bolouri, M. Mayorga, A. Hamberger, Neuropathology and pressure in the pig brain resulting from low-impulse noise exposure. J. Neurotrauma 25, 1397–1406 (2008).
- S. Knudsen, E. O. Øen, Blast-induced neurotrauma in whales. Neurosci. Res. 46, 377–386 (2003).
- J. B. Long, T. L. Bentley, K. A. Wessner, C. Cerone, S. Sweeney, R. A. Bauman, Blast overpressure in rats: Recreating a battlefield injury in the laboratory. J. Neurotrauma 26, 827–840 (2009).
- R. A. Bauman, G. Ling, L. Tong, A. Januszkiewicz, D. Agoston, N. Delanerolle, Y. Kim, D. Ritzel, R. Bell, J. Ecklund, R. Armonda, F. Bandak, S. Parks, An introductory characterization of a combat-casualty-care relevant swine model of closed head injury resulting from exposure to explosive blast. J. Neurotrauma 26, 841–860 (2009).
- S. I. Svetlov, V. Prima, D. R. Kirk, H. Gutierrez, K. C. Curley, R. L. Hayes, K. K. Wang, Morphologic and biochemical characterization of brain injury in a model of controlled blast overpressure exposure. J. Trauma 69, 795–804 (2010).
- I. Cernak, A. C. Merkle, V. E. Koliatsos, J. M. Bilik, Q. T. Luong, T. M. Mahota, L. Xu, N. Slack, D. Windle, F. A. Ahmed, The pathobiology of blast injuries and blast-induced neurotrauma as identified using a new experimental model of injury in mice. Neurobiol. Dis. 41, 538–551 (2011).
- V. E. Koliatsos, I. Cernak, L. Xu, Y. Song, A. Savonenko, B. J. Crain, C. G. Eberhart, C. E. Frangakis, T. Melnikova, H. Kim, D. Lee, A mouse model of blast injury to brain: Initial pathological, neuropathological, and behavioral characterization. J. Neuropathol. Exp. Neurol. 70, 399–416 (2011).
- M. Chavko, T. Watanabe, S. Adeeb, J. Lankasky, S. T. Ahlers, R. M. McCarron, Relationship between orientation to a blast and pressure wave propagation inside the rat brain. J. Neurosci. Methods 195, 61–66 (2011).
- A. Nakagawa, G. T. Manley, A. D. Gean, K. Ohtani, R. Armonda, A. Tsukamoto, H. Yamamoto, K. Takayama, T. Tominaga, Mechanisms of primary blast-induced traumatic brain injury: Insights from shock-wave research. J. Neurotrauma 28, 1101–1119 (2011).
- M. Risling, S. Plantman, M. Angeria, E. Rostami, B. M. Bellander, M. Kirkegaard, U. Arborelius, J. Davidsson, Mechanisms of blast induced brain injuries, experimental studies in rats. Neuroimage 54 (Suppl. 1), S89–S97 (2011).
- J. Lu, K. C. Ng, G. Ling, J. Wu, D. J. Poon, E. M. Kan, M. H. Tan, Y. J. Wu, P. Li, S. Moochhala, E. Yap, L. K. Lee, M. Teo, I. B. Yeh, D. M. Sergio, F. Chua, S. D. Kumar, E. A. Ling, Effect of blast exposure on the brain structure and cognition in Macaca fascicularis. J. Neurotrauma 29, 1434–1454 (2012).
- F. W. Mott, The effects of high explosives upon the central nervous system. Lancet 187, 331–338, 441–449, 545–553 (1916). War Neuroses and Shell Shock (Frowde, Hodder &Stoughton, London, 1919).
- J. F. Fulton, Blast and concussion in the present war. N. Engl. J. Med. 226, 1–8 (1942).
- D. Denny-Brown, The sequelae of war head injuries. N. Engl. J. Med. 227, 771–780 (1942).
- H. Cohen, G. R. Biskind, Pathologic aspects of atmospheric blast injuries in man. Arch. Pathol. 42, 12–34 (1946).
- G. A. Elder, E. M. Mitsis, S. T. Ahlers, A. Cristian, Blast-induced mild traumatic brain injury. Psychiatr. Clin. North Am. 33, 757–781 (2010).
- B. Omalu, J. L. Hammers, J. Bailes, R. L. Hamilton, M. I. Kamboh, G. Webster, R. P. Fitzsimmons, Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg. Focus 31, E3 (2011).
- D. W. Hyde, CONWEP 2.1.0.8, Conventional Weapons Effects Program (United States Army Corps of Engineers, Vicksburg, MS, 2004).
- T. J. Nelson, T. Clark, E. T. Stedje-Larsen, C. T. Lewis, J. M. Grueskin, E. L. Echols, D. B. Wall, E. A. Felger, H. R. Bohman, Close proximity blast injury patterns from improvised explosive devices in Iraq: A report of 18 cases. J. Trauma 65, 212–217 (2008).
- The Jackson Laboratory, MPD: Jaxpheno1. Mouse Phenome Database; http://phenome.jax.org [accessed 22 February 2012].
- D. T. Blackstock, in Fundamentals of Physical Acoustics (Wiley, New York, 2000), pp. 146–150.
- R. J. Cloots, J. A. van Dommelen, T. Nyberg, S. Kleiven, M. G. Geers, Micromechanics of diffuse axonal injury: Influence of axonal orientation and anisotropy. Biomech. Model. Mechanobiol. 10, 413–422 (2011).
- H. T. Tran, F. M. Laferla, D. M. Holtzman, D. L. Brody, Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-b accumulation and independently accelerates the development of tau abnormalities. J. Neurosci. 31, 9513–9525 (2011).
- Y. Shitaka, H. T. Tran, R. E. Bennett, L. Sanchez, M. A. Levy, K. Dikranian, D. L. Brody, Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J. Neuropathol. Exp. Neurol. 70, 551–567 (2011).
- K. Hynynen, N. McDannold, H. Martin, F. A. Jolesz, N. Vykhodtseva, The threshold for brain damage in rabbits induced by bursts of ultrasound in the presence of an ultrasound contrast agent (Optison). Ultrasound Med. Biol. 29, 473–481 (2003).
- A. P. Evan, L. R. Willis, J. A. McAteer, M. R. Bailey, B. A. Connors, Y. Shao, J. E. Lingeman, J. C. Williams Jr., N. S. Fineberg, L. A. Crum, Kidney damage and renal functional changes are minimized by waveform control that suppresses cavitation in shock wave lithotripsy. J. Urol. 168, 1556–1562 (2002).
- L. A. Crum, Y. Mao, Acoustically enhanced bubble growth at low frequencies and its implications for human diver and marine mammal safety. J. Acoust. Soc. Am. 99, 2898–2907 (1996).
- C. J. Clemedson, H. Pettersson, Propagation of a high explosive air shock wave through different parts of an animal body. Am. J. Physiol. 184, 119–126 (1956).
- M. Chavko, W. A. Koller, W. K. Prusaczyk, R. M. McCarron, Measurement of blast wave by a miniature fiber optic pressure transducer in the rat brain. J. Neurosci. Methods 159, 277–281 (2007).
- A. H. S. Holbourn, Mechanics of head injuries. Lancet 242, 438–441 (1943).
- S. J. Strich, Shearing of nerve fibres as a cause of brain damage due to head injury: A pathological study of twenty cases. Lancet 2, 443–448 (1961).
- T. A. Gennarelli, L. E. Thibault, A. K. Ommaya, in Biomechanics of Impact Injury and Injury Tolerances of the Head-Neck Complex, S. H. Backaitis, Ed. (Society of Automotive Engineers Inc., Warrendale, PA, 1993).
- U. Krave, S. Höjer, H. A. Hansson, Transient, powerful pressures are generated in the brain by a rotational acceleration impulse to the head. Eur. J. Neurosci. 21, 2876–2882 (2005).
- J. Zhang, N. Yoganandan, F. A. Pintar, T. A. Gennarelli, Role of translational and rotational accelerations on brain strain in lateral head impact. Biomed. Sci. Instrum. 42, 501–506 (2006).
- J. T. Povlishock, Pathobiology of traumatically induced axonal injury in animals and man. Ann. Emerg. Med. 22, 980–986 (1993).
- R. J. Cloots, H. M. Gervaise, J. A. van Dommelen, M. G. Geers, Biomechanics of traumatic brain injury: Influences of the morphologic heterogeneities of the cerebral cortex. Ann. Biomed. Eng. 36, 1203–1215 (2008).
- W. C. Moss, M. J. King, E. G. Blackman, Skull flexure from blast waves: A mechanism for brain injury with implications for helmet design. Phys. Rev. Lett. 103, 108702 (2009).
- R. D. Bell, E. A. Winkler, A. P. Sagare, I. Singh, B. LaRue, R. Deane, B. V. Zlokovic, Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68, 409–427 (2010).
- U. Ito, Y. Hakamata, E. Kawakami, K. Oyanagi, Temporary cerebral ischemia results in swollen astrocytic end-feet that compress microvessels and lead to delayed focal cortical infarction. J. Cereb. Blood Flow Metab. 31, 328–338 (2011).
- R. J. Fijalkowski, B. D. Stemper, F. A. Pintar, N. Yoganandan, M. J. Crowe, T. A. Gennarelli, New rat model for diffuse brain injury using coronal plane angular acceleration. J. Neurotrauma 24, 1387–1398 (2007).
- P. W. Alford, B. E. Dabiri, J. A. Goss, M. A. Hemphill, M. D. Brigham, K. K. Parker, Blast-induced phenotypic switching in cerebral vasospasm. Proc. Natl. Acad. Sci. U.S.A. 108, 12705–12710 (2011).
- K. D. Browne, X. H. Chen, D. F. Meaney, D. H. Smith, Mild traumatic brain injury and diffuse axonal injury in swine. J. Neurotrauma 28, 1747–1755 (2011).
- F. Zhu, H. Mao, A. Dal Cengio Leonardi, C. Wagner, C. Chou, X. Jin, C. Bir, P. Vandevord, K. H. Yang, A. I. King, Development of an FE model of the rat head subjected to air shock loading. Stapp Car Crash J. 54, 211–225 (2010).
- R. J. Kelleher III, A. Govindarajan, S. Tonegawa, Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron 44, 59–73 (2004).
- P. Tsokas, E. A. Grace, P. Chan, T. Ma, S. C. Sealfon, R. Iyengar, E. M. Landau, R. D. Blitzer, Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J. Neurosci. 25, 5833–5843 (2005).
- C. R. Bramham, Local protein synthesis, actin dynamics, and LTP consolidation. Curr. Opin. Neurobiol. 18, 524–531 (2008).
- G. Barkhoudarian, D. A. Hovda, C. C. Giza, The molecular pathophysiology of concussive brain injury. Clin. Sports Med. 30, 33–48 (2011).
- E. J. Pellman, D. C. Viano, A. M. Tucker, I. R. Casson, J. F. Waeckerle, Concussion in professional football: Reconstruction of game impacts and injuries. Neurosurgery 53, 799–812 (2003).
- K. M. Guskiewicz, J. P. Mihalik, V. Shankar, S. W. Marshall, D. H. Crowell, S. M. Oliaro, M. F. Ciocca, D. N. Hooker, Measurement of head impacts in collegiate football players: Relationship between head impact biomechanics and acute clinical outcome after concussion. Neurosurgery 61, 1244–1252 (2007).
- S. P. Broglio, B. Schnebel, J. J. Sosnoff, S. Shin, X. Fend, X. He, J. Zimmerman, Biomechanical properties of concussions in high school football. Med. Sci. Sports Exerc. 42, 2064–2071 (2010).
- T. R. Terrell, R. M. Bostick, R. Abramson, D. Xie, W. Barfield, R. Cantu, M. Stanek, T. Ewing, APOE, APOE promoter, and Tau genotypes and risk for concussion in college athletes. Clin. J. Sport Med. 18, 10–17 (2008).
- J. J. Vasterling, S. P. Proctor, P. Amoroso, R. Kane, T. Heeren, R. F. White, Neuropsychological outcomes of army personnel following deployment to the Iraq war. JAMA 296, 519–529 (2006).
- M. B. Stein, T. W. McAllister, Exploring the convergence of posttraumatic stress disorder and mild traumatic brain injury. Am. J. Psychiatry 166, 768–776 (2009).
- J. J. Vasterling, S. P. Proctor, M. J. Friedman, C. W. Hoge, T. Heeren, L. A. King, D. W. King, PTSD symptom increases in Iraq-deployed soldiers: Comparison with nondeployed soldiers and associations with baseline symptoms, deployment experiences, and postdeployment stress. J. Trauma Stress 23, 41–51 (2010).
- J. P. Vonsattel, H. Aizawa, P. Ge, M. DiFiglia, A. C. McKee, M. MacDonald, J. F. Gusella, G. B. Landwehrmeyer, E. D. Bird, E. P. Richardson Jr., E. T. Hedley-Whyte, An improved approach to prepare human brains for research. J. Neuropathol. Exp. Neurol. 54, 42–56 (1995).
- C. A. Barnes, Memory deficits associated with senescence: A neurophysiological and behavioral study in the rat. J. Comp. Physiol. Psychol. 93, 74–104 (1979).
- S. Saman, W. Kim, M. Raya, Y. Visnick, S. Miro, S. Saman, B. Jackson, A. C. McKee, V. E. Alvarez, N. C. Lee, G. F. Hall, Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849 (2012).
- S. A. Formby, R. K. Wharton, Blast characteristics and TNT equivalence values for some commercial explosives detonated at ground level. J. Hazard. Mater. 50, 183–198 (1996).
- I. G. Cullis, Blast waves and how they interact with structures. J. R. Army Med. Corps 147, 16–26 (2001).
- H. R. Champion, J. B. Holcomb, L. A. Young, Injuries from explosions: Physics, biophysics, pathology, and required research focus. J. Trauma 66, 1468–1477 (2009).
- Acknowledgments: We gratefully acknowledge the use of resources and facilities at the EdithNourse Rogers Memorial Veterans Hospital (Bedford, MA) and expert assistance provided by A. Peters (Department of Anatomy and Neurobiology, Boston University School of Medicine) for electron microscopy consultation, L. McHale and M. Nowinski for tireless logistical assistance and administrative support, K. Cormier and C. A. Kubilus for histotechnology assistance, and P. Davies (Albert Einstein College of Medicine) and J. Bertelsen (Signet/Covance) for antibodies. We gratefully acknowledge the individuals and families whose participation and contributions made this work possible. This work is dedicated to U.S. military servicemen and women, past and present, for their sacrifices, dedication, and service to our country. Funding: Supported by National Institute of General Medical Sciences grant GM75986 (L.E.G.); National Center for Research Resources grant S10RR026599 (L.E.G.); NSF grants 0901760 and 0821304 (L.E.G.); Cure Alzheimer’s Fund (L.E.G., R.D.M., and R.E.T.), Office of the Dean, Boston University School of Medicine; Office of the Provost, Boston University (L.E.G.); Boston University Photonics Center Neurophotonics Pilot Project Grant (L.E.G.); Cooperative Agreement W911NG- 06-2-0040 between the U.S. Army and Boston University Photonics Center (L.E.G.); Department of Veterans Affairs (A.C.M.); Translational Research Center for TBI and Stress Disorders (TRACTS) VA Rehabilitation Research and Development Traumatic Brain Injury Center of Excellence (B6796-C; A.C.M., L.E.G.); National Institute of Aging Center grant P30AG13846 (N.W.K.), Supplement 0572063345-5 (N.W.K.), and Pilot Project Grant (N.C.); NIH/National Institute of Biomedical Imaging and Bioengineering T32 Training Grant Award 5T32EB006359 and NIH/National Institute of Neurological Disorders and Stroke Pre-Doctoral Fellowship Research grant F31NS077796 (C.A.T.); National Institute of Diabetes and Digestive and Kidney Diseases grant P01DK043881 (R.O.C.); National Institute of Neurological Diseases and Stroke grants NS044421 (P.K.S.), NS072966 (L.V.), and R01NS078337 (R.A.S.); Department of Defense grant W81XWH (P.K.S.); Migraine Research Foundation (P.K.S.); March of Dimes Foundation grant 6FY08591 (L.V.); National Operating Committee on Standards for Athletic Equipment (A.C.M.); an unrestricted gift from the National Football League (A.C.M.); and Department of Energy Contract DE-AC52-07NA27344 to the Lawrence Livermore National Laboratory (W.C.M.). Author contributions: L.E.G. designed and interpreted all experiments, analyzed electron microscopy data, designed the shock tube apparatus, provided funding, and wrote the paper; A.M.F., C.A.T., M.W.W., J.M.K., N.C., J.A.M., and O.M. performed and analyzed all shock tube blast mouse experiments and harvested brain tissue; X.-L.Z. designed and performed all electrophysiology experiments; L.V. designed, analyzed, and interpreted all behavioral experiments, provided funding, and wrote the paper; J.A.S. designed and performed electrophysiology and behavioral experiments; C.U. designed and performed electrophysiology experiments; M.E. performed electron microscopy; C.J.G. and G.M.M. performed and analyzed behavioral experiments; R.D.M., S.S., and G.F.H. designed, performed, and interpreted tau biochemical analyses; C.J.N., R.A.S., R.C.C., and J.G. provided and interpreted human clinical data; J.K.B., B.L.W., T.I., A.E.B., and N.W.K. designed animal experiments and edited the paper; T.D.S. analyzed neuropathological data; D.C. and A.S. designed the shock tube blast apparatus; W.C.M. interpreted biophysical data from shock tube experiments and wrote and edited the paper; R.O.C. designed and interpreted all biophysical data from shock tube experiments and wrote and edited the paper; R.E.T. designed and interpreted all experiments and wrote and edited the paper; P.K.S. designed and interpreted electrophysiology and behavioral experiments, provided funding, and wrote the paper; A.C.M. designed and interpreted human clinical and neuropathological data, provided funding, and wrote the paper. Competing interests: G.F.H. is a Science Advisory Board member of Immunotrex Biologics Inc. The other authors declare that they have no competing interests. The views expressed in this article are those of authors J.G., A.E.B., N.W.K., and A.C.M. and should not to be construed as official positions of the Department of Veterans Affairs or the U.S. government. Portions of this document were prepared, in part, as an account of work by W.C.M. sponsored by an agency of the U.S. government. Neither the U.S. government nor Lawrence Livermore National Security, LLC, nor any of their employees makes any warranty, expressed or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the U.S. government or Lawrence Livermore National Security, LLC. The views and opinions of author W.C.M. expressed herein do not necessarily state or reflect those of the U.S. government or Lawrence Livermore National Security, LLC, and shall not be used for advertising or product endorsement purposes.

